Opening requested calculation...
Please wait, this takes like 47 seconds. Thank you for your patience! :)
☠
0 humans have been terminated by curable diseases since this page started loading
For every 1 unit of harm prevented by efficacy delays, the system creates 3,389 (95% CI: 1,811-5,734) units of harm through denied access. Operational safety ratio: 3,389 (95% CI: 1,811-5,734)-to-1 against the patient.
In 1962, your Congress decided that after proving a drug won’t kill you, sick people should wait another 8.2 years (95% CI: 4.84 years-11.5 years) before they’re allowed to take it. The drug passed safety testing. It won’t kill you, won’t melt your liver, won’t make your hair migrate to your back. But you can’t have it, because a committee needs to spend 8.2 years (95% CI: 4.84 years-11.5 years) making sure it works well enough. I asked someone why, and they said “we need to confirm efficacy.” I said, “The patient is dying.” They said, “Yes, but we don’t know if the drug works.” I said, “The patient could tell you, if you gave it to them.” They said that’s not how it works. I said, “Dying is how it works?” They changed the subject, which I suppose is also how it works.
You’re not allowed to volunteer for the trials that would answer the question faster, either. You are, however, allowed to die. Nobody blocks that. There’s no 8.2 years (95% CI: 4.84 years-11.5 years) waiting period on dying.
Your species confirms something won’t harm you, then locks it in a cabinet and forms a committee. The committee meets quarterly. You decompose on a different schedule.
A Note on Blame
When this chapter says “the FDA does X,” read it as “the 1962 law requires the FDA to do X.” Congress wrote the rules. The FDA follows them. Blaming the FDA is like blaming the gun instead of the person who loaded it, aimed it at patients, and pulled the trigger on a 62-year schedule.
“But What About Safety?”
This is the part where humans panic, so let me be precise: keep Phase I safety testing. It works. It caught thalidomide. Nobody is suggesting you skip it. Do radically more efficacy testing, on real patients, with transparent results. But stop holding known-safe drugs hostage while you run the trials.
There is no word for “preventing someone from accessing a safe substance that could help them while they die.” No sane civilization would do it, so none needed a word. You have several words for it. My favorite is “the approval process.”
Compare two real-world systems for testing drugs:
- The Oxford RECOVERY Trial: Tested COVID treatments on 48,000 patients for $500 (95% CI: $400-$2,500). Found a life-saving treatment in 3 months. Like a normal species would.
- The Post-1962 System: Averages $41,000 (95% CI: $20,000-$120,000) per trial. Takes over a decade182 to approve a new drug. Like humans would.
Your efficacy testing system costs 44.1x (95% CI: 12.8x-210x) more and takes 32.8x (95% CI: 19.4x-45.9x) longer than necessary. I showed these numbers to a human and they said “that can’t be right.” So I showed them the sources and they said “well, there must be a reason.” There is a reason. The reason is that your system costs 44.1x (95% CI: 12.8x-210x) more and takes 32.8x (95% CI: 19.4x-45.9x) longer. That’s the reason and also the problem. They’re the same thing.
Here’s where your lifespan goes to die:
- Phase I (Safety Testing): 2.3 years - Tests if the drug harms people: death, organ damage, mutations, serious adverse reactions, all the ways a molecule can ruin your day. This part works. Keep it.
- Phase II/III (Efficacy Testing): 8.2 years (95% CI: 4.84 years-11.5 years) - The drug is now known safe. But patients can’t have it until the committee is satisfied. This is where people die waiting for a drug that already passed every safety check your species invented.
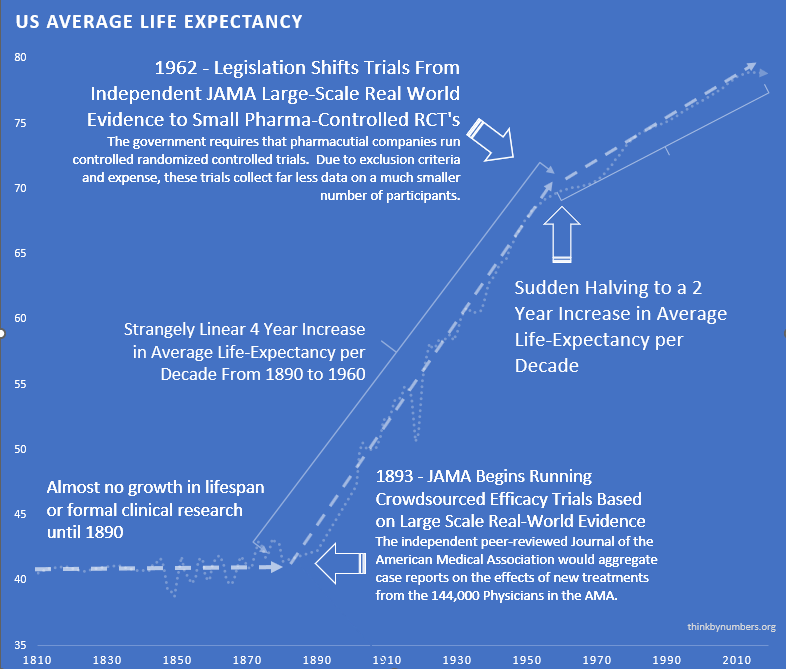
What the 1962 Efficacy Requirements Changed
Before 1962, 144 thousand physicians tested treatments on real patients and published results. Life expectancy grew 3.82 years every decade163 for 80 straight years. Things were going well. Naturally, Congress intervened.
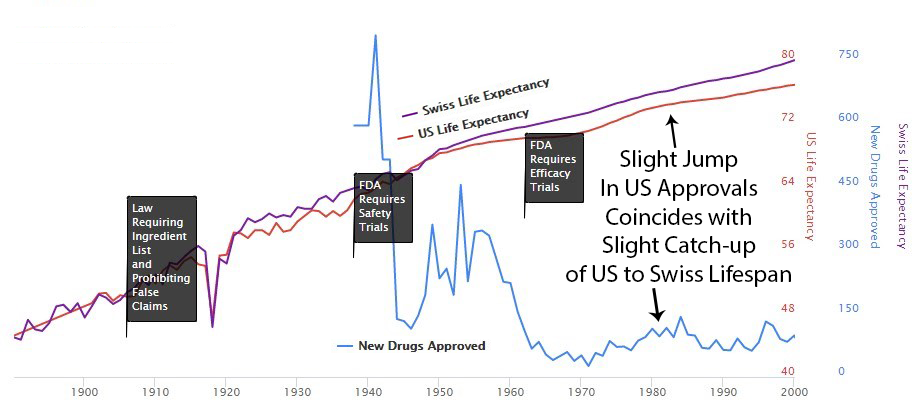
Phase I safety testing (added in 1938) prevented all US thalidomide deaths while Europe had thousands. So in 1962, Congress looked at a system that was working and thought, “What if we made this much worse?” They added massive pre-approval efficacy requirements via the Kefauver Harris Amendment183, blocking patient access for years during testing.
The cost of developing a drug went from $24.7 million (95% CI: $19.5 million-$30 million) to $2.6 billion (95% CI: $1.5 billion-$4 billion), a 105x (95% CI: 72.8x-149x) increase. New drug approvals dropped by 70%124. Life expectancy gains slowed from 3.82 years per decade to 1.54163,164, a 60% reduction. Time to approval stretched from 2-3 years to 10.5 years (95% CI: 6 years-12 years).
Everything got worse at exactly the same time. Every single metric, all at once, the moment the law passed. I’m not a statistician so I can’t tell you whether that’s a coincidence, but I did look at the graph, and it’s not a coincidence.
The 105x (95% CI: 72.8x-149x) cost increase is rigorously documented. See Drug Development Cost Analysis for methodology, sensitivity analysis, and source data.
The Modern Consequences
Even if you think historical analysis is just opinions with footnotes, the current system’s failures are happening right now:
- A cure passes safety testing
- You can’t take it (pending efficacy review)
- You can’t join the trial either (too sick, too old, wrong medications)
- You die
- The FDA approves the cure
- Pharmaceutical companies charge your widow $10,000 per pill
I read this sequence several times because I kept thinking I’d got the order wrong. I hadn’t. That is the correct order. The system works exactly like that, on purpose, and everyone involved considers it a success because nobody skipped a step.
High Costs Kill Innovation, Reward Monopoly
Before 1962: Genius scientist invents cure, raises a few million, tests safety, gives it to people. Like a civilization that wants to survive would.
Now: Genius scientist must convince one of three mega-corporations to fund trials. Those corporations already sell inferior drugs for the same condition, bringing in hundreds of millions a year per drug.
The math for the mega-corporation:
Option 1: Develop the better drug
Cost: $2.6 billion (95% CI: $1.5 billion-$4 billion) on average to get a single drug to market. That figure already includes the cost of the nine candidates that fail for every one that succeeds, so the 10% success rate is already priced in.
Best case, the drug works: you now have a better drug for the same condition. But your old drug, which was bringing in hundreds of millions a year, is now obsolete. You spent $2.6 billion (95% CI: $1.5 billion-$4 billion) to replace your own revenue stream. Every year the new drug is on the market, it’s earning roughly what the old drug would have earned anyway, minus the $2.6 billion (95% CI: $1.5 billion-$4 billion) hole you dug getting here.
Option 2: Buy the patent, put it on a shelf
Your inferior drug keeps bringing in hundreds of millions a year. Total cost: whatever the patent costs, which is rounding error compared to $2.6 billion (95% CI: $1.5 billion-$4 billion).
I asked a pharmaceutical executive which option they’d choose and they said “it’s more nuanced than that.” Then I asked which one they chose and they said Option 2. So it wasn’t more nuanced than that.
The profit incentive doesn’t just fail to reward better treatments. It actively punishes them. The best possible outcome of Option 1 is the same annual revenue you already had, minus $2.6 billion (95% CI: $1.5 billion-$4 billion). Curing disease is economically irrational. The system doesn’t need a conspiracy to suppress cures. It just needs the math. I keep waiting for someone to show me where the math is wrong. Nobody shows me where the math is wrong. They just look uncomfortable and say “it’s complicated,” which I’ve learned is what humans say when it isn’t complicated and they don’t like the answer.
Off-Patent Drugs and Rare Diseases: Mathematically Doomed
The 1962 law made it mathematically impossible to cure diseases that aren’t profitable enough:
- 95% of diseases are rare127: Development cost ($2.6 billion (95% CI: $1.5 billion-$4 billion)) / patient population (~10,000 patients) = $260,000/patient
- No patent = no funding: Off-patent drugs can’t attract billion-dollar investments
- Pre-specification kills serendipity: Must predict what drug cures before testing
When something costs more, you get less of it. I looked this up and apparently it’s the first thing they teach in economics. It is also, somehow, the last thing anyone considers when writing health policy. 95% of diseases127 have zero treatments. Not because cures are impossible. Because cures are unprofitable. The best thing a disease can do for itself, career-wise, is be popular.
The Actual Death Toll of “Drug Lag”
Economists call it “drug lag,” which is a lovely, clinical way of saying “people rotting to death in a queue for drugs that have already been confirmed not to hurt them.” Early estimates: 21,000 to 120,000 American lives per decade184. But that was just the US.
A full accounting using WHO mortality data puts the total cost of the 8.2 years (95% CI: 4.84 years-11.5 years) post-safety efficacy delay (the lag after drugs are known safe) at 416 million deaths (95% CI: 244 million deaths-587 million deaths) eventually avoidable deaths.
Including years lived with disability: 8.77 billion DALYs (95% CI: 4.88 billion DALYs-13.2 billion DALYs) of healthy human life deleted. Economic cost: $1.32 quadrillion (95% CI: $676 trillion-$2.14 quadrillion) (2024 USD), reflecting those 8.77 billion DALYs (95% CI: 4.88 billion DALYs-13.2 billion DALYs) valued at $150,000 (95% CI: $100,384-$198,679)/DALY, the standard normative rate used in WHO cost-effectiveness analyses.
Even if those death estimates are off by a factor of 10, that’s still tens of millions of deaths. And the inefficiency is independently verifiable: the efficacy testing process costs 82x (95% CI: 21.4x-195x) more per patient and takes 32.8x (95% CI: 19.4x-45.9x) longer than proven alternatives like the Oxford RECOVERY trial. Those numbers don’t depend on mortality estimates at all.
Here’s a news story from the Non-Existent Times by No One Ever, without a picture of all the people who die from lack of access to life-saving treatments that might have been. You can’t photograph millions of people who died from not receiving a drug. There’s no body with a toe tag reading “cause of death: regulatory delay.” They die of cancer, heart disease, Alzheimer’s. The delay that killed them is invisible, so nobody writes the headline.
Types of Error in FDA Approval Decision
| FDA Allows the Drug |
Correct Decision |
Victims appear on television. Regulator is fired. |
| FDA Does Not Allow the Drug |
Victims die at home. Nobody is fired. |
Correct Decision |
So the FDA has two kinds of mistakes. One where they give you a bad drug and you die on television. And one where they don’t give you a good drug and you die at home. They’ve decided the second one doesn’t count, which is convenient because it’s the one they do most.
The most infamous case: beta-blockers. Europe used them to prevent heart attacks. The US delayed for a decade, killing an estimated 100,000 Americans185. More than died in Vietnam and Korea combined. From one drug. That they had. That they knew worked. That they put in a drawer. I asked someone if there was a word for killing 100,000 people by not doing something, and they said “it depends on the context.” I think 100,000 dead people is the context, but apparently that’s not enough context.
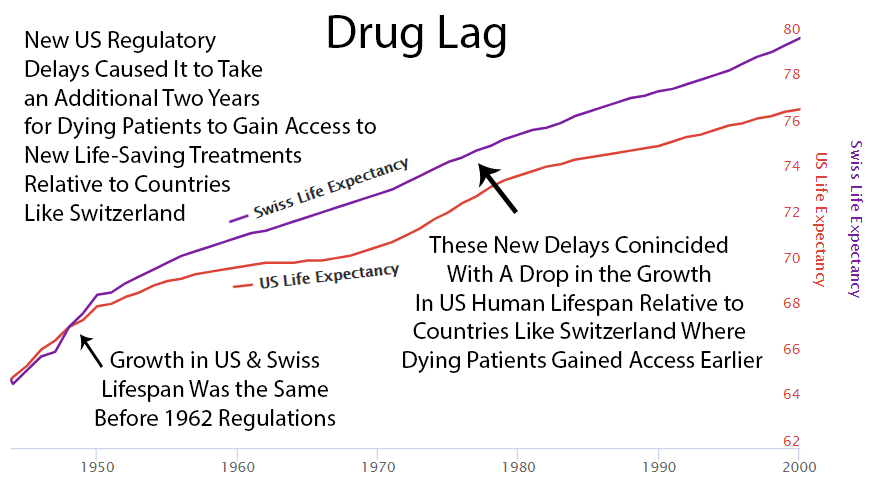
After 1962, US life expectancy diverged from Switzerland’s (which didn’t introduce the same delays). More drug approvals in the ’80s narrowed the gap. Fewer approvals in the ’90s widened it again. The pattern is not subtle. I showed it to a child and the child understood it. I showed it to a policy expert and they said it was “multifactorial.” I think “multifactorial” is what adults say when they can see the same thing the child can see but have reasons not to say it out loud.
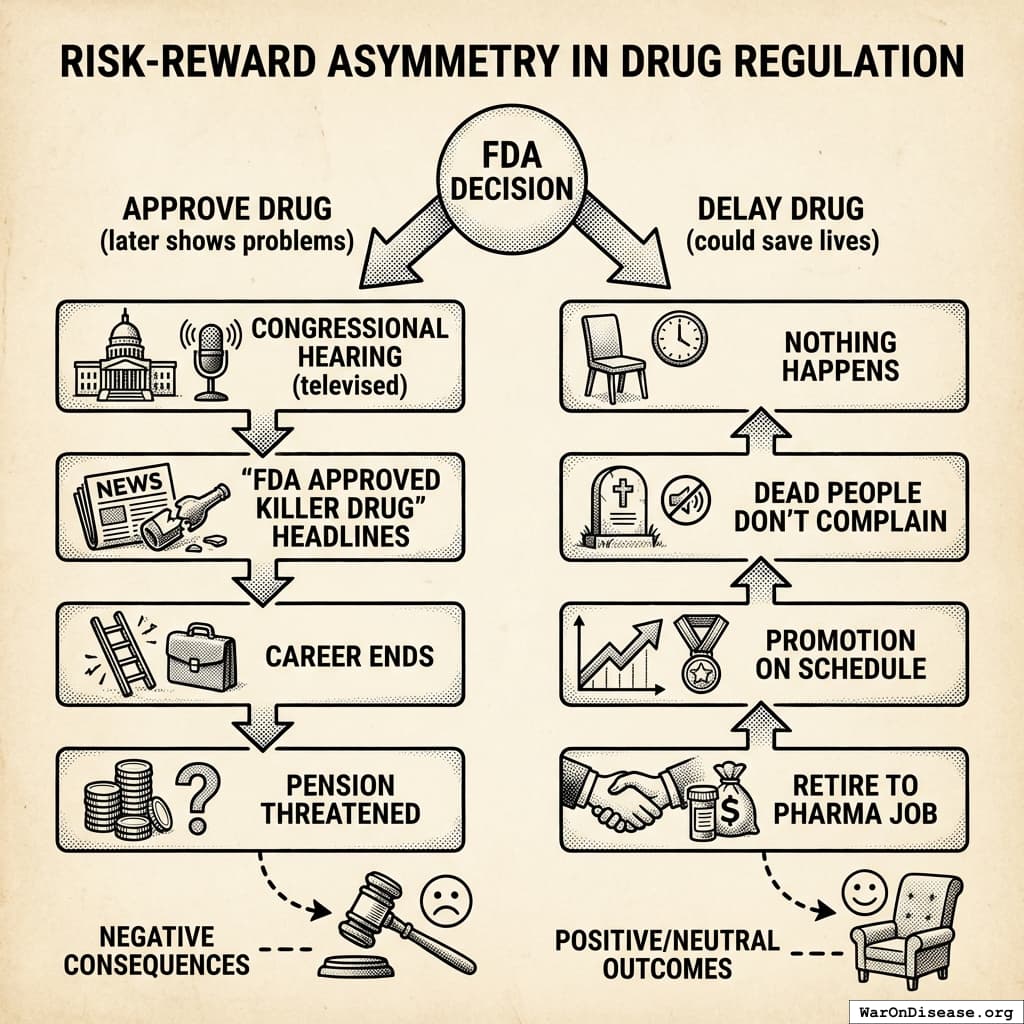
How the Incentives Work
FDA Regulator Decision Tree
Approve drug that later shows problems
- Congressional hearing (televised)
- “FDA APPROVED KILLER DRUG” headlines
- Career ends
- Pension threatened
Delay drug that could save lives
- Nothing happens
- Nobody writes a headline
- Promotion on schedule
- Retire to pharma job186
To be fair, this isn’t a conspiracy. It’s just a system where the only career-ending move is letting patients access treatments. If you approve a bad drug, CNN says your name. If you delay a good drug, nobody dies on television. So the rational move is to delay everything, and that’s what happens, and everyone who could object is dead, which does simplify the feedback process.
Dr. Henry I. Miller ran the FDA team reviewing recombinant human insulin in the early 1980s. Mountains of evidence showed it was safe and effective. His supervisor refused to approve it. If someone died from the drug, heads would roll. If people died waiting for the drug? Nothing happens.
Dead patients don’t testify before Congress. I checked.
The Math: Why Current Regulations Increase Total Harm
\[
\begin{gathered}
Ratio_{TypeII} \\
= \frac{DALYs_{lag}}{DALY_{TypeI}} \\
= \frac{8.77B}{2.59M} \\
= 3{,}390
\end{gathered}
\]
where:
\[
DALYs_{lag} = YLL_{lag} + YLD_{lag} = 7.9B + 873M = 8.77B
\]
where:
\[
\begin{gathered}
YLL_{lag} \\
= \text{DEATHS\_TOTAL} \times (REMAINING_LIFE_EXPECTANCY_AT_60 - (\text{MEAN\_AGE\_OF\_DEATH} - 60))
\end{gathered}
\]
where:
\[
\begin{gathered}
Deaths_{lag} \\
= T_{lag} \times Deaths_{disease,daily} \times 338 \\
= 8.2 \times 150{,}000 \times 338 \\
= 416M
\end{gathered}
\]
where:
\[
\begin{gathered}
YLD_{lag} \\
= Deaths_{lag} \times T_{suffering} \times DW_{chronic} \\
= 416M \times 6 \times 0.35 \\
= 873M
\end{gathered}
\]
where:
\[
\begin{gathered}
DALY_{TypeI} \\
= DALY_{thal} \times 62 \\
= 41{,}800 \times 62 \\
= 2.59M
\end{gathered}
\]
where:
\[
\begin{gathered}
DALY_{thal} \\
= YLD_{thal} + YLL_{thal} \\
= 13{,}000 + 28{,}800 \\
= 41{,}800
\end{gathered}
\]
where:
\[
\begin{gathered}
YLD_{thal} \\
= DW_{thal} \times N_{thal,survive} \times LE_{thal} \\
= 0.4 \times 540 \times 60 \\
= 13{,}000
\end{gathered}
\]
where:
\[
\begin{gathered}
N_{thal,survive} \\
= N_{thal,US,prevent} \times (1 - Rate_{thal,mort}) \\
= 900 \times (1 - 40\%) \\
= 540
\end{gathered}
\]
where:
\[
\begin{gathered}
N_{thal,US,prevent} \\
= N_{thal,global} \times Pct_{US,1960} \\
= 15{,}000 \times 6\% \\
= 900
\end{gathered}
\]
where:
\[
\begin{gathered}
YLL_{thal} \\
= Deaths_{thal} \times 80 \\
= 360 \times 80 \\
= 28{,}800
\end{gathered}
\]
where:
\[
\begin{gathered}
Deaths_{thal} \\
= Rate_{thal,mort} \times N_{thal,US,prevent} \\
= 40\% \times 900 \\
= 360
\end{gathered}
\]
Even generously crediting ALL drug withdrawals to the 1962 changes (ignoring that Phase I safety testing already existed), the prevented harm totals ~2.59 million DALYs (95% CI: 1.88 million DALYs-3.38 million DALYs). The delay harm totals 8.77 billion DALYs (95% CI: 4.88 billion DALYs-13.2 billion DALYs).
For every 1 unit of harm prevented, the current system creates 3,389 (95% CI: 1,811-5,734) units of harm through delay. The safety net catches one person and drops 3,389 (95% CI: 1,811-5,734). There is a word for that. The word is “hole.”
Clinical Trial Theater: Excluding 86.1% of Reality Makes Drugs More Dangerous
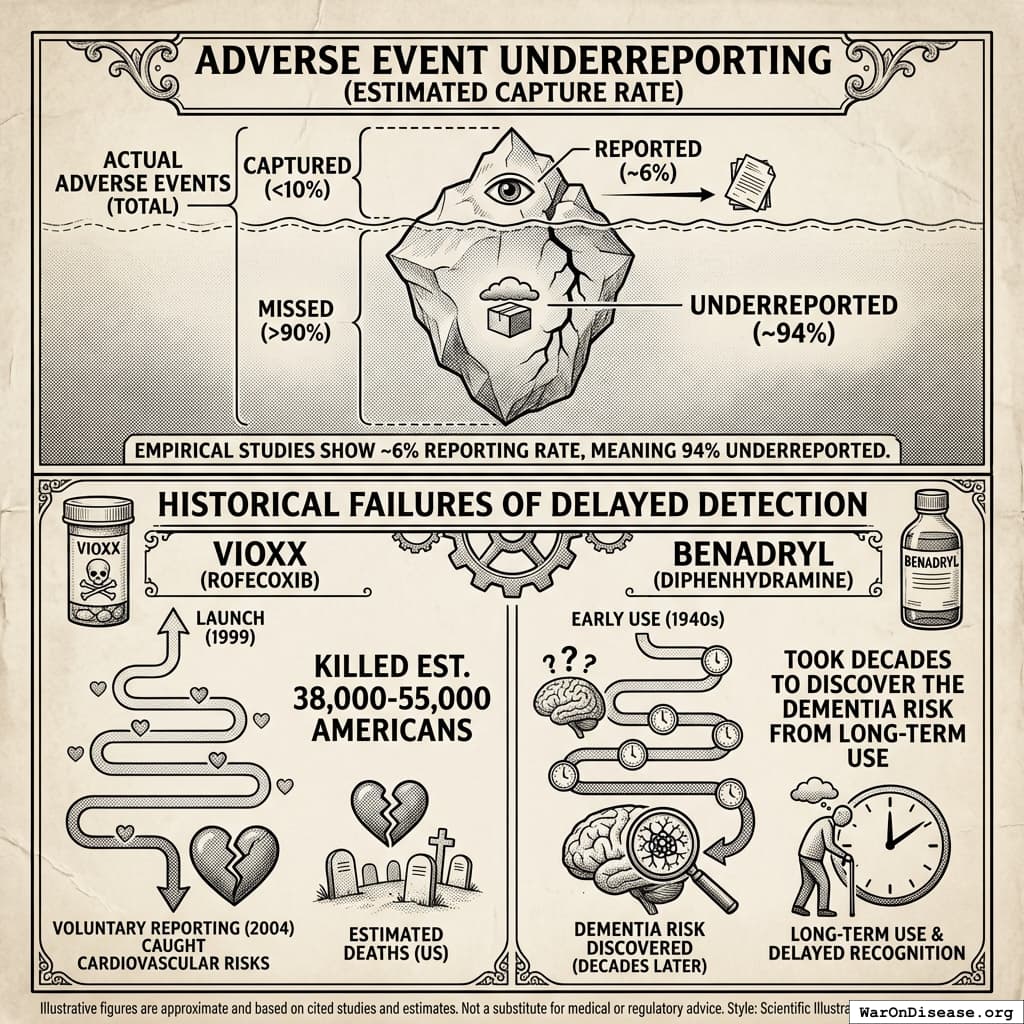
To be clear: Phase I safety testing should exclude vulnerable populations. You don’t give an untested molecule to a pregnant woman. But once a drug clears safety, the efficacy trials that follow also exclude those populations, which means nobody learns whether the drug works for the people who will actually take it, or what interactions to expect. Then the drug is approved for everyone, and the post-market surveillance system (FAERS) catches less than 10% of adverse events. So the exclusions create a data gap, and nothing ever fills it.
The FDA requires rigorous safety trials. Good. Then it runs efficacy trials on people who aren’t sick, aren’t old, aren’t on other drugs, and aren’t pregnant. I read this three times. They test whether drugs work on people who don’t need the drugs, to predict whether they’ll work for people who do need the drugs, who are different people, with different bodies, taking different medications. Then they act surprised when the second group has a reaction. I would call this stupid but I think it might actually be something worse than stupid. Stupid doesn’t usually have a peer-review process.
Trials under the current system exclude5:
- Patients over 65: Most people who actually take medications (excluded due to “comorbidities”)
- Patients under 18: All children (metabolism differs from adults)
- Pregnant women: Excluded entirely (then drugs prescribed during pregnancy anyway)
- Anyone with comorbidities: The sickest patients most likely to have adverse reactions
- Anyone on other medications: Everyone elderly (can’t detect drug interactions)
- Anyone too far from trial sites: Poor and rural populations
It’s like testing whether a car is safe by driving it very slowly into a nice cushion, and then selling it to people who drive into walls. And when they die you say “well, it was safe when WE tried it.” Which is true. It was.
Result: In antidepressant trials alone, 86.1% of real patients are excluded. The drugs are tested on the healthiest people alive, declared “safe and effective,” then handed to everyone the trial specifically avoided. Nobody knows whether the drug works for those populations, what the interactions are, or what the real adverse event profile looks like. This is how Vioxx, Fen-Phen, and Bextra all got approved, celebrated, prescribed to millions, and then withdrawn after killing people whose demographics the trials had carefully excluded. The 8.2 years (95% CI: 4.84 years-11.5 years) delay didn’t make these drugs safer. It made them seem safer by hiding the danger behind an exclusion list.
Testing drugs only on unrepresentative volunteers to prove they work is like testing parachutes only on the ground. The parachute worked perfectly, by the way. It just wasn’t falling at the time.
No Long-Term Outcome Data
Data collection can be as short as several months. You take the drug for forty years. The gap between these two numbers is where side effects live. Nobody checks. It’s like reviewing a marriage based on the wedding.
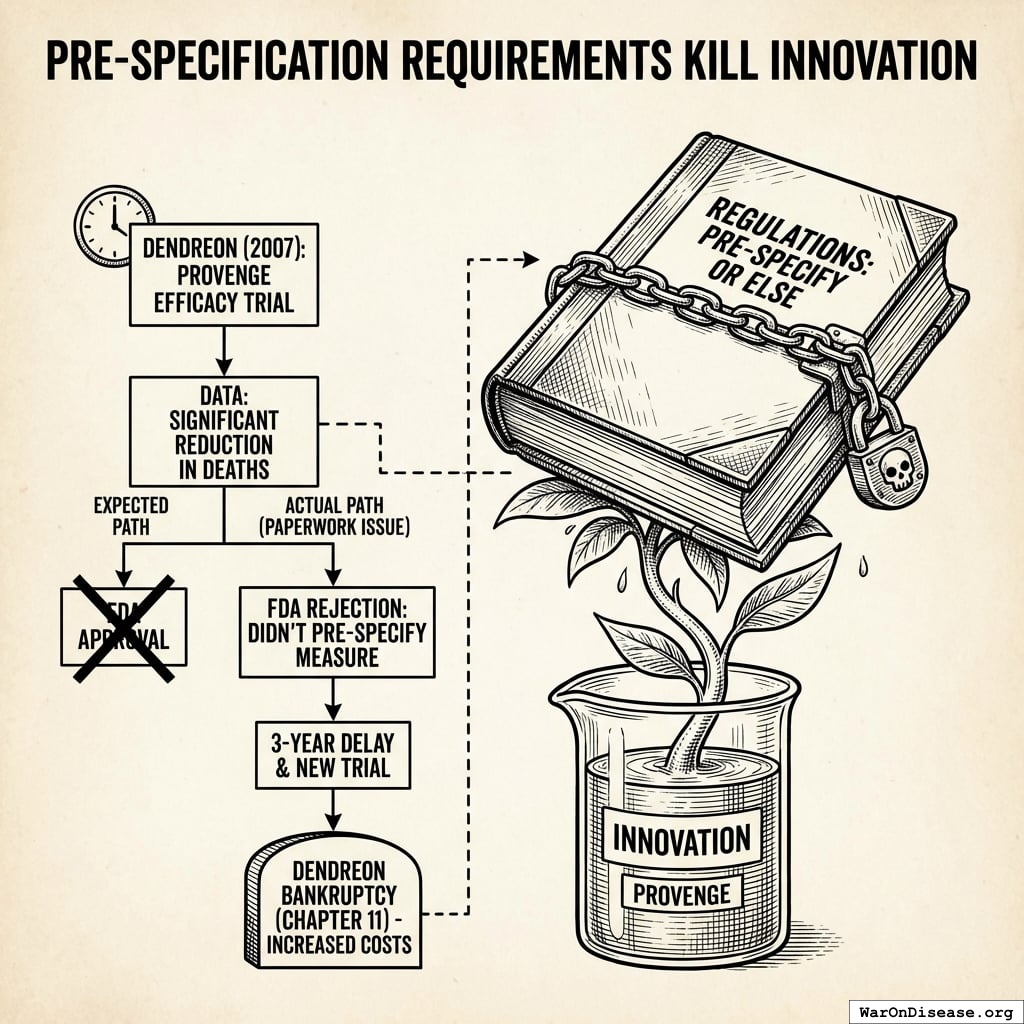
Pre-Specification Requirements Kill Innovation
The regulations require drug developers to predict exactly what a treatment will cure before testing it on humans. You must know the answer before you’re allowed to ask the question. I asked if this was how science works and they said “it’s how regulatory science works,” which I think means “no, but we gave it the same name.”
In 2007, Dendreon showed that its immunotherapy drug Provenge significantly reduced deaths from prostate cancer. The FDA advisory committee agreed it worked. Then the FDA rejected it anyway. Not because it didn’t save lives. Because Dendreon filled out the wrong form about which lives it was planning to save.
“People stopped dying” wasn’t good enough. The paperwork predicting that people would stop dying wasn’t filed in the right order. Three more years and another trial were required. During those three years, people who could have stopped dying continued dying, but at least the forms were correct. A rational civilization would have a saying for this: “The gravestone was filed on time.” It means someone prioritized procedure over the thing the procedure was supposed to protect. You could use it daily.
Due to these additional costs, Dendreon filed for chapter 11 bankruptcy187. So the approval process killed the patients, then killed the company trying to save them. You have a phrase for this: “killing two birds with one stone.” Although in this case the stone was paperwork and there were considerably more than two birds. But you can’t count the dead birds who would have been cured by a company you bankrupted.
The Blinding Catch-22: How Knowing a Drug Works Proves It Doesn’t
MDMA was synthesized in 1912188. By the 1970s, therapists were using it to treat trauma, and an estimated half a million doses were administered in therapeutic settings189. In 1985, the government scheduled it. In 1986, Rick Doblin founded MAPS to get it approved for PTSD190. Thirty-eight years and over $140 million in philanthropic funding later, they ran two successful Phase 3 trials. 67% of participants no longer met the diagnostic criteria for PTSD191. The drug worked. Veterans with treatment-resistant PTSD got better. The FDA’s own advisory committee reviewed the data.
They voted 9-2 that it wasn’t effective192.
The problem: patients could tell they’d taken MDMA, because MDMA makes you feel things, which is how it treats PTSD, which is the point. About 90% of participants in the drug group correctly guessed they weren’t on placebo. The FDA called this “functional unblinding” and said it invalidated the results193.
I want to make sure I understand the logic. The drug works by making you feel profoundly different. The FDA requires that you not know whether you feel profoundly different. If you notice that you feel profoundly different, the trial fails, because you weren’t supposed to notice. The drug’s mechanism of action is, itself, a protocol violation.
This is not a fixable design problem. You cannot double-blind a drug whose therapeutic effect is that the patient feels it working. The FDA knows this. They granted MDMA “Breakthrough Therapy Designation” in 2017, signaling that yes, this looks promising, please spend another seven years and tens of millions of dollars on Phase 3 trials. Then they rejected it for a property of the molecule that was evident before the trials began. No sane inspector would tell someone to build a house and then condemn it for having walls. Here, you owe them nothing. The veterans kept their PTSD. The FDA kept its process. Everyone kept exactly what they had, which was the problem in the first place.
Meanwhile, approximately 49,000 Americans kill themselves every year194, including 6,400 veterans195 at twice the civilian rate. About 13 million Americans have PTSD in any given year196, and PTSD nearly quadruples your suicide risk197. MDMA-assisted therapy produced a 67% remission rate versus 32% for standard care. Even conservatively, if half of PTSD patients had accessed the treatment over the 40 years since it was scheduled, the 35-percentage-point improvement in remission would have prevented something on the order of 30,000 suicides in the US alone, and freed millions of people from a condition that destroys families, drives substance abuse, and fills your homeless shelters with veterans.
A molecule that Merck synthesized the same year the Titanic sank is sitting in regulatory limbo because the people it helped could tell it was helping them. I looked for the word that describes letting thousands of people kill themselves each year while a known-effective treatment waits for someone to solve an unsolvable blinding problem, and the closest I found was “process.”
Small Trials Are Dangerous
Phase III trials test 1,000-3,000 patients. A 1-in-10,000 adverse event is mathematically invisible in a trial of 3,000. Then the drug goes to millions.
After approval, the FDA monitors safety using FAERS, a system where doctors voluntarily report problems if they feel like it. It captures less than 10% of actual adverse events198. Vioxx killed an estimated 38,000-55,000 Americans199 before this honor system noticed. That’s more people than died in the entire Korean War, detected by a system that relies on doctors filling out optional paperwork. I want to be clear: the 8.2 years (95% CI: 4.84 years-11.5 years) delay is supposed to be for safety. And then the safety monitoring after approval is a suggestion box. You waited 8.2 years (95% CI: 4.84 years-11.5 years) to get to the suggestion box.
A decentralized FDA200,201 with 10 million participants catches 1-in-10,000 reactions, includes real populations, tracks long-term outcomes, and provides automated surveillance instead of voluntary paperwork. You have a fax machine and a suggestion box. One of these is a safety system. The other is a prayer with a 8.2 years (95% CI: 4.84 years-11.5 years) waiting period.
The Negative Results Black Hole
Here’s something that would be illegal if anybody important cared:
- Negative trial results published: 37%202
- Positive trial results published: 94%202
- Money wasted repeating failed experiments: ~$100 billion annually203
Companies are allowed to hide when drugs don’t work. Restaurants can’t hide when they fail a health inspection. I checked this twice because I thought I was wrong, but I wasn’t. A sandwich has more regulatory transparency than chemotherapy. I don’t know what to do with that information but I’ve had it for three days now and it won’t go away.
Company: “We tested this drug!”
Regulator: “Did it work?”
Company: “…We tested this drug!”
Pharmaceutical companies bury negative results deeper than Jimmy Hoffa. Other companies waste billions testing the same dead ends. Your insurance premiums fund this magnificent inefficiency. It’s like casinos only having to report when people win. Actually, I think casinos DO have to report when people win. So drugs get less honesty than gambling. I keep finding these comparisons and they keep getting worse.
Countries That Don’t Have Our “Safety”
Japan204 gives conditional approval after Phase II with real-world data collection. Time to patient: 2-3 years. Americans fly there for treatment.
The EU205 lets terminal patients access experimental drugs at a doctor’s discretion. I asked the FDA what might go wrong and they said “the patient could die.” The patient is already dying. “Yes,” they said, “but they could die differently.” I waited for the rest of the sentence but that was it.
Right to Try (US)206 passed despite FDA opposition. The FDA made it effectively impossible to use. The law says you can ask a manufacturer for their experimental drug. The manufacturer can say no. They almost always say no, because the law provides no liability protection, no funding mechanism, and no insurance coverage. So “Right to Try” means “Right to Ask a Corporation for a Favor While Dying.” Corporations said no. Patients helped: <200 total207. Patients who wanted help: tens of thousands. Everywhere else in the universe, “Right to Try” is just called “rights.” The fact that your species needed a special law to allow dying people to take known-safe drugs, and then made the law effectively unusable, is the most human thing I’ve ever observed. You named it “Right to Try.” The name was accurate, which was so unusual for your species that it should have been suspicious. And it was: the law doesn’t work. The naming convention holds.
The COVID Test Fiasco
January 2020: WHO develops COVID test, world starts using it. February: FDA blocks all non-CDC tests208 to “ensure quality.” The CDC tests were contaminated209. Private labs begged to help. Late March: FDA finally allows other tests after thousands die210.
The only approved tests didn’t work. The unapproved ones did. The FDA blocked the ones that worked to ensure the quality of the ones that didn’t. I’ve written this paragraph four times now and it keeps sounding like I’m making it up, but I’m not. That is what happened. In order. On purpose.
The Bottom Line
To be fair to the FDA, the system does eventually approve most good drugs. So if you think about it, the system works. Unless you were one of the people who died waiting, in which case the system worked slightly less well for you specifically. But you’re dead, so you can’t complain, which means the system has a 100% satisfaction rate among living participants. That’s actually quite impressive if you don’t think about it.
It is not a safety system. It is a filing system that produces corpses as a byproduct.
The only thing missing is the political will to admit that a law signed before the Beatles existed might not be the pinnacle of pharmaceutical regulation. The Beatles broke up. The law didn’t. Draw your own conclusions.
P.S. - The FDA will likely object to this chapter. Estimated response time: 12-15 years, pending Phase III review and proper documentation. The full quantitative model is at invisible-graveyard.warondisease.org, for those who enjoy numbers large enough to induce nausea.
1.
NIH Common Fund. NIH pragmatic trials: Minimal funding despite 30x cost advantage.
NIH Common Fund: HCS Research Collaboratory https://commonfund.nih.gov/hcscollaboratory (2025)
The NIH Pragmatic Trials Collaboratory funds trials at $500K for planning phase, $1M/year for implementation-a tiny fraction of NIH’s budget. The ADAPTABLE trial cost $14 million for 15,076 patients (= $929/patient) versus $420 million for a similar traditional RCT (30x cheaper), yet pragmatic trials remain severely underfunded. PCORnet infrastructure enables real-world trials embedded in healthcare systems, but receives minimal support compared to basic research funding. Additional sources: https://commonfund.nih.gov/hcscollaboratory | https://pcornet.org/wp-content/uploads/2025/08/ADAPTABLE_Lay_Summary_21JUL2025.pdf | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5604499/
.
4.
Cato Institute. Chance of dying from terrorism statistic.
Cato Institute: Terrorism and Immigration Risk Analysis https://www.cato.org/policy-analysis/terrorism-immigration-risk-analysis Chance of American dying in foreign-born terrorist attack: 1 in 3.6 million per year (1975-2015) Including 9/11 deaths; annual murder rate is 253x higher than terrorism death rate More likely to die from lightning strike than foreign terrorism Note: Comprehensive 41-year study shows terrorism risk is extremely low compared to everyday dangers Additional sources: https://www.cato.org/policy-analysis/terrorism-immigration-risk-analysis | https://www.nbcnews.com/news/us-news/you-re-more-likely-die-choking-be-killed-foreign-terrorists-n715141
.
5.
NIH. Antidepressant clinical trial exclusion rates.
Zimmerman et al. https://pubmed.ncbi.nlm.nih.gov/26276679/ (2015)
Mean exclusion rate: 86.1% across 158 antidepressant efficacy trials (range: 44.4% to 99.8%) More than 82% of real-world depression patients would be ineligible for antidepressant registration trials Exclusion rates increased over time: 91.4% (2010-2014) vs. 83.8% (1995-2009) Most common exclusions: comorbid psychiatric disorders, age restrictions, insufficient depression severity, medical conditions Emergency psychiatry patients: only 3.3% eligible (96.7% excluded) when applying 9 common exclusion criteria Only a minority of depressed patients seen in clinical practice are likely to be eligible for most AETs Note: Generalizability of antidepressant trials has decreased over time, with increasingly stringent exclusion criteria eliminating patients who would actually use the drugs in clinical practice Additional sources: https://pubmed.ncbi.nlm.nih.gov/26276679/ | https://pubmed.ncbi.nlm.nih.gov/26164052/ | https://www.wolterskluwer.com/en/news/antidepressant-trials-exclude-most-real-world-patients-with-depression
.
7.
CNBC. Warren buffett’s career average investment return.
CNBC https://www.cnbc.com/2025/05/05/warren-buffetts-return-tally-after-60-years-5502284percent.html (2025)
Berkshire’s compounded annual return from 1965 through 2024 was 19.9%, nearly double the 10.4% recorded by the S&P 500. Berkshire shares skyrocketed 5,502,284% compared to the S&P 500’s 39,054% rise during that period. Additional sources: https://www.cnbc.com/2025/05/05/warren-buffetts-return-tally-after-60-years-5502284percent.html | https://www.slickcharts.com/berkshire-hathaway/returns
.
8.
World Health Organization. WHO global health estimates 2024.
World Health Organization https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates (2024)
Comprehensive mortality and morbidity data by cause, age, sex, country, and year Global mortality: 55-60 million deaths annually Lives saved by modern medicine (vaccines, cardiovascular drugs, oncology): 12M annually (conservative aggregate) Leading causes of death: Cardiovascular disease (17.9M), Cancer (10.3M), Respiratory disease (4.0M) Note: Baseline data for regulatory mortality analysis. Conservative estimate of pharmaceutical impact based on WHO immunization data (4.5M/year from vaccines) + cardiovascular interventions (3.3M/year) + oncology (1.5M/year) + other therapies. Additional sources: https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates
.
9.
GiveWell. GiveWell cost per life saved for top charities (2024).
GiveWell: Top Charities https://www.givewell.org/charities/top-charities General range: $3,000-$5,500 per life saved (GiveWell top charities) Helen Keller International (Vitamin A): $3,500 average (2022-2024); varies $1,000-$8,500 by country Against Malaria Foundation: $5,500 per life saved New Incentives (vaccination incentives): $4,500 per life saved Malaria Consortium (seasonal malaria chemoprevention): $3,500 per life saved VAS program details: $2 to provide vitamin A supplements to child for one year Note: Figures accurate for 2024. Helen Keller VAS program has wide country variation ($1K-$8.5K) but $3,500 is accurate average. Among most cost-effective interventions globally Additional sources: https://www.givewell.org/charities/top-charities | https://www.givewell.org/charities/helen-keller-international | https://ourworldindata.org/cost-effectiveness
.
11.
U.S. Department of Defense.
5.56mm NATO ammunition bulk procurement pricing. (2024)
The cost of 5.56mm NATO ammunition at military bulk procurement rates is approximately $0.40 per round, based on Lake City Army Ammunition Plant production and commercial market floor prices for mil-spec M855 ammunition.
12.
Pike, J.
U.s. Forces fire 250,000 rounds for every insurgent killed. (2011)
The General Accounting Office reports that US forces used 1.8 billion rounds of small-arms ammunition per year, a level that more than doubled in five years. An estimated 250,000 rounds were fired for every insurgent killed in Iraq and Afghanistan.
13.
AARP. Unpaid caregiver hours and economic value.
AARP 2023 https://www.aarp.org/caregiving/financial-legal/info-2023/unpaid-caregivers-provide-billions-in-care.html (2023)
Average family caregiver: 25-26 hours per week (100-104 hours per month) 38 million caregivers providing 36 billion hours of care annually Economic value: $16.59 per hour = $600 billion total annual value (2021) 28% of people provided eldercare on a given day, averaging 3.9 hours when providing care Caregivers living with care recipient: 37.4 hours per week Caregivers not living with recipient: 23.7 hours per week Note: Disease-related caregiving is subset of total; includes elderly care, disability care, and child care Additional sources: https://www.aarp.org/caregiving/financial-legal/info-2023/unpaid-caregivers-provide-billions-in-care.html | https://www.bls.gov/news.release/elcare.nr0.htm | https://www.caregiver.org/resource/caregiver-statistics-demographics/
.
15.
Forbes.
Forbes world’s billionaires list 2024. (2024)
Forbes identified a record 2,781 billionaires worldwide with combined net worth of $14.2 trillion, 141 more than 2023. Bernard Arnault (LVMH) topped the list at $233 billion.
16.
CDC MMWR. Childhood vaccination economic benefits.
CDC MMWR https://www.cdc.gov/mmwr/volumes/73/wr/mm7331a2.htm (1994)
US programs (1994-2023): $540B direct savings, $2.7T societal savings ( $18B/year direct, $90B/year societal) Global (2001-2020): $820B value for 10 diseases in 73 countries ( $41B/year) ROI: $11 return per $1 invested Measles vaccination alone saved 93.7M lives (61% of 154M total) over 50 years (1974-2024) Additional sources: https://www.cdc.gov/mmwr/volumes/73/wr/mm7331a2.htm | https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(24)00850-X/fulltext
.
20.
U.S. Bureau of Labor Statistics.
CPI inflation calculator. (2024)
CPI-U (1980): 82.4 CPI-U (2024): 313.5 Inflation multiplier (1980-2024): 3.80× Cumulative inflation: 280.48% Average annual inflation rate: 3.08% Note: Official U.S. government inflation data using Consumer Price Index for All Urban Consumers (CPI-U). Additional sources: https://www.bls.gov/data/inflation_calculator.htm
.
21.
James Surowiecki.
The Wisdom of Crowds. (Surowiecki, 2004).
Explores the aggregation of information in groups, arguing that decisions are often better than could have been made by any single member of the group. The opening anecdote relates Francis Galton’s surprise that the crowd at a county fair accurately guessed the weight of an ox when the median of their individual guesses was taken. The three conditions for a group to be intelligent are diversity, independence, and decentralization. Additional sources: https://archive.org/details/wisdomofcrowds0000suro | https://en.wikipedia.org/wiki/The_Wisdom_of_Crowds | https://www.amazon.com/Wisdom-Crowds-James-Surowiecki/dp/0385721706
.
22.
ClinicalTrials.gov API v2 direct analysis. ClinicalTrials.gov cumulative enrollment data (2025).
Direct analysis via ClinicalTrials.gov API v2 https://clinicaltrials.gov/data-api/api Analysis of 100,000 active/recruiting/completed trials on ClinicalTrials.gov (as of January 2025) shows cumulative enrollment of 12.2 million participants: Phase 1 (722k), Phase 2 (2.2M), Phase 3 (6.5M), Phase 4 (2.7M). Median participants per trial: Phase 1 (33), Phase 2 (60), Phase 3 (237), Phase 4 (90). Additional sources: https://clinicaltrials.gov/data-api/api
.
23.
ACS CAN. Clinical trial patient participation rate.
ACS CAN: Barriers to Clinical Trial Enrollment https://www.fightcancer.org/policy-resources/barriers-patient-enrollment-therapeutic-clinical-trials-cancer Only 3-5% of adult cancer patients in US receive treatment within clinical trials About 5% of American adults have ever participated in any clinical trial Oncology: 2-3% of all oncology patients participate Contrast: 50-60% enrollment for pediatric cancer trials (<15 years old) Note: 20% of cancer trials fail due to insufficient enrollment; 11% of research sites enroll zero patients Additional sources: https://www.fightcancer.org/policy-resources/barriers-patient-enrollment-therapeutic-clinical-trials-cancer | https://hints.cancer.gov/docs/Briefs/HINTS_Brief_48.pdf
.
24.
ScienceDaily. Global prevalence of chronic disease.
ScienceDaily: GBD 2015 Study https://www.sciencedaily.com/releases/2015/06/150608081753.htm (2015)
2.3 billion individuals had more than five ailments (2013) Chronic conditions caused 74% of all deaths worldwide (2019), up from 67% (2010) Approximately 1 in 3 adults suffer from multiple chronic conditions (MCCs) Risk factor exposures: 2B exposed to biomass fuel, 1B to air pollution, 1B smokers Projected economic cost: $47 trillion by 2030 Note: 2.3B with 5+ ailments is more accurate than "2B with chronic disease." One-third of all adults globally have multiple chronic conditions Additional sources: https://www.sciencedaily.com/releases/2015/06/150608081753.htm | https://pmc.ncbi.nlm.nih.gov/articles/PMC10830426/ | https://pmc.ncbi.nlm.nih.gov/articles/PMC6214883/
.
25.
C&EN. Annual number of new drugs approved globally: 50.
C&EN https://cen.acs.org/pharmaceuticals/50-new-drugs-received-FDA/103/i2 (2025)
50 new drugs approved annually Additional sources: https://cen.acs.org/pharmaceuticals/50-new-drugs-received-FDA/103/i2 | https://www.fda.gov/drugs/development-approval-process-drugs/novel-drug-approvals-fda
.
26.
Williams, R. J., Tse, T., DiPiazza, K. & Zarin, D. A.
Terminated trials in the ClinicalTrials.gov results database: Evaluation of availability of primary outcome data and reasons for termination.
PLOS One 10, e0127242 (2015)
Approximately 12% of trials with results posted on the ClinicalTrials.gov results database (905/7,646) were terminated. Primary reasons: insufficient accrual (57% of non-data-driven terminations), business/strategic reasons, and efficacy/toxicity findings (21% data-driven terminations).
29.
OpenSecrets. Defense sector lobbying summary.
OpenSecrets https://www.opensecrets.org/federal-lobbying/sectors/summary?id=D (2025)
Military sector federal lobbying totaled $198,009,793 in 2025, up from $159.5 million in 2024 and $142.9 million in 2023. Additional sources: https://www.opensecrets.org/federal-lobbying/sectors/summary?id=D
.
30.
Companies Market Cap.
BAE systems and thales market capitalization. (2026)
BAE Systems market capitalization approx $75.80B and Thales approx $56.68B as of June 2026, combined approx $132.5B for the two major allied European military primes. Additional sources: https://companiesmarketcap.com/thales/marketcap/
.
31.
Stock Analysis.
Military prime contractor market capitalization and float statistics. (2026)
Combined market capitalization of 11 US military primes approx $835.8B at the 2026-06-11 close: RTX $248.07B, Boeing $174.71B, Lockheed Martin $126.51B, General Dynamics $96.90B, Northrop Grumman $78.48B, L3Harris $58.16B, Leidos $15.36B, Huntington Ingalls $11.86B, CACI $11.61B, Booz Allen Hamilton $9.24B, SAIC $4.86B. Tradeable float across the 13 Western primes (adding BAE Systems and Thales) approx $880B, about 91 percent of combined cap (range $850-900B), from per-company float and shares-outstanding statistics pages; big-5 floats verified individually (RTX 92.6%, BA 96.0%, LMT 85.7%, GD 94.2%, NOC 99.7%); Thales is the outlier at approx 45% float because the French State (26.60%) and Dassault Aviation (26.59%) stakes are locked. Additional sources: https://stockanalysis.com/stocks/rtx/statistics/ | https://www.dassault-aviation.com/en/group/about-us/shareholding-structure-and-organization-chart/
.
32.
Rummel, R. J.
Death by Government: Genocide and Mass Murder Since 1900. (Transaction Publishers, 1994).
Political scientist R.J. Rummel’s comprehensive accounting of democide (government murder of unarmed civilians) in the 20th century. His final revised estimate: 262 million people murdered by their own governments from 1900-1999, excluding battle deaths in wars. Range: 200-272+ million. Communist regimes account for the largest share (100-148+ million). Updated figures at hawaii.edu/powerkills.
33.
GiveWell. Cost per DALY for deworming programs.
https://www.givewell.org/international/technical/programs/deworming/cost-effectiveness Schistosomiasis treatment: $28.19-$70.48 per DALY (using arithmetic means with varying disability weights) Soil-transmitted helminths (STH) treatment: $82.54 per DALY (midpoint estimate) Note: GiveWell explicitly states this 2011 analysis is "out of date" and their current methodology focuses on long-term income effects rather than short-term health DALYs Additional sources: https://www.givewell.org/international/technical/programs/deworming/cost-effectiveness
.
35.
Calculated from IHME Global Burden of Disease (2.55B DALYs) and global GDP per capita valuation. $109 trillion annual global disease burden.
The global economic burden of disease, including direct healthcare costs ($8.2 trillion) and lost productivity ($100.9 trillion from 2.55 billion DALYs × $39,570 per DALY), totals approximately $109.1 trillion annually.
37.
Think by Numbers. Pre-1962 drug development costs and timeline (think by numbers).
Think by Numbers: How Many Lives Does FDA Save? https://thinkbynumbers.org/health/how-many-net-lives-does-the-fda-save/ (1962)
Historical estimates (1970-1985): USD $226M fully capitalized (2011 prices) 1980s drugs: $65M after-tax R&D (1990 dollars), $194M compounded to approval (1990 dollars) Modern comparison: $2-3B costs, 7-12 years (dramatic increase from pre-1962) Context: 1962 regulatory clampdown reduced new treatment production by 70%, dramatically increasing development timelines and costs Note: Secondary source; less reliable than Congressional testimony Additional sources: https://thinkbynumbers.org/health/how-many-net-lives-does-the-fda-save/ | https://en.wikipedia.org/wiki/Cost_of_drug_development | https://www.statnews.com/2018/10/01/changing-1962-law-slash-drug-prices/
.
38.
Biotechnology Innovation Organization (BIO). BIO clinical development success rates 2011-2020.
Biotechnology Innovation Organization (BIO) https://go.bio.org/rs/490-EHZ-999/images/ClinicalDevelopmentSuccessRates2011_2020.pdf (2021)
Phase I duration: 2.3 years average Total time to market (Phase I-III + approval): 10.5 years average Phase transition success rates: Phase I→II: 63.2%, Phase II→III: 30.7%, Phase III→Approval: 58.1% Overall probability of approval from Phase I: 12% Note: Largest publicly available study of clinical trial success rates. Efficacy lag = 10.5 - 2.3 = 8.2 years post-safety verification. Additional sources: https://go.bio.org/rs/490-EHZ-999/images/ClinicalDevelopmentSuccessRates2011_2020.pdf
.
39.
Nature Medicine. Drug repurposing rate ( 30%).
Nature Medicine https://www.nature.com/articles/s41591-024-03233-x (2024)
Approximately 30% of drugs gain at least one new indication after initial approval. Additional sources: https://www.nature.com/articles/s41591-024-03233-x
.
40.
EPI. Education investment economic multiplier (2.1).
EPI: Public Investments Outside Core Infrastructure https://www.epi.org/publication/bp348-public-investments-outside-core-infrastructure/ Early childhood education: Benefits 12X outlays by 2050; $8.70 per dollar over lifetime Educational facilities: $1 spent → $1.50 economic returns Energy efficiency comparison: 2-to-1 benefit-to-cost ratio (McKinsey) Private return to schooling: 9% per additional year (World Bank meta-analysis) Note: 2.1 multiplier aligns with benefit-to-cost ratios for educational infrastructure/energy efficiency. Early childhood education shows much higher returns (12X by 2050) Additional sources: https://www.epi.org/publication/bp348-public-investments-outside-core-infrastructure/ | https://documents1.worldbank.org/curated/en/442521523465644318/pdf/WPS8402.pdf | https://freopp.org/whitepapers/establishing-a-practical-return-on-investment-framework-for-education-and-skills-development-to-expand-economic-opportunity/
.
41.
PMC. Healthcare investment economic multiplier (1.8).
PMC: California Universal Health Care https://pmc.ncbi.nlm.nih.gov/articles/PMC5954824/ (2022)
Healthcare fiscal multiplier: 4.3 (95% CI: 2.5-6.1) during pre-recession period (1995-2007) Overall government spending multiplier: 1.61 (95% CI: 1.37-1.86) Why healthcare has high multipliers: No effect on trade deficits (spending stays domestic); improves productivity & competitiveness; enhances long-run potential output Gender-sensitive fiscal spending (health & care economy) produces substantial positive growth impacts Note: "1.8" appears to be conservative estimate; research shows healthcare multipliers of 4.3 Additional sources: https://pmc.ncbi.nlm.nih.gov/articles/PMC5954824/ | https://cepr.org/voxeu/columns/government-investment-and-fiscal-stimulus | https://ncbi.nlm.nih.gov/pmc/articles/PMC3849102/ | https://set.odi.org/wp-content/uploads/2022/01/Fiscal-multipliers-review.pdf
.
42.
World Bank. Infrastructure investment economic multiplier (1.6).
World Bank: Infrastructure Investment as Stimulus https://blogs.worldbank.org/en/ppps/effectiveness-infrastructure-investment-fiscal-stimulus-what-weve-learned (2022)
Infrastructure fiscal multiplier: 1.6 during contractionary phase of economic cycle Average across all economic states: 1.5 (meaning $1 of public investment → $1.50 of economic activity) Time horizon: 0.8 within 1 year, 1.5 within 2-5 years Range of estimates: 1.5-2.0 (following 2008 financial crisis & American Recovery Act) Italian public construction: 1.5-1.9 multiplier US ARRA: 0.4-2.2 range (differential impacts by program type) Economic Policy Institute: Uses 1.6 for infrastructure spending (middle range of estimates) Note: Public investment less likely to crowd out private activity during recessions; particularly effective when monetary policy loose with near-zero rates Additional sources: https://blogs.worldbank.org/en/ppps/effectiveness-infrastructure-investment-fiscal-stimulus-what-weve-learned | https://www.gihub.org/infrastructure-monitor/insights/fiscal-multiplier-effect-of-infrastructure-investment/ | https://cepr.org/voxeu/columns/government-investment-and-fiscal-stimulus | https://www.richmondfed.org/publications/research/economic_brief/2022/eb_22-04
.
43.
Mercatus. Military spending economic multiplier (0.6).
Mercatus: Defense Spending and Economy https://www.mercatus.org/research/research-papers/defense-spending-and-economy Ramey (2011): 0.6 short-run multiplier Barro (1981): 0.6 multiplier for WWII spending (war spending crowded out 40¢ private economic activity per federal dollar) Barro & Redlick (2011): 0.4 within current year, 0.6 over two years; increased govt spending reduces private-sector GDP portions General finding: $1 increase in deficit-financed federal military spending = less than $1 increase in GDP Variation by context: Central/Eastern European NATO: 0.6 on impact, 1.5-1.6 in years 2-3, gradual fall to zero Ramey & Zubairy (2018): Cumulative 1% GDP increase in military expenditure raises GDP by 0.7% Additional sources: https://www.mercatus.org/research/research-papers/defense-spending-and-economy | https://cepr.org/voxeu/columns/world-war-ii-america-spending-deficits-multipliers-and-sacrifice | https://www.rand.org/content/dam/rand/pubs/research_reports/RRA700/RRA739-2/RAND_RRA739-2.pdf
.
48.
FDA. FDA-approved prescription drug products (20,000+).
FDA https://www.fda.gov/media/143704/download There are over 20,000 prescription drug products approved for marketing. Additional sources: https://www.fda.gov/media/143704/download
.
53.
ACLED. Active combat deaths annually.
ACLED: Global Conflict Surged 2024 https://acleddata.com/2024/12/12/data-shows-global-conflict-surged-in-2024-the-washington-post/ (2024)
2024: 233,597 deaths (30% increase from 179,099 in 2023) Deadliest conflicts: Ukraine (67,000), Palestine (35,000) Nearly 200,000 acts of violence (25% higher than 2023, double from 5 years ago) One in six people globally live in conflict-affected areas Additional sources: https://acleddata.com/2024/12/12/data-shows-global-conflict-surged-in-2024-the-washington-post/ | https://acleddata.com/media-citation/data-shows-global-conflict-surged-2024-washington-post | https://acleddata.com/conflict-index/index-january-2024/
.
54.
UCDP. State violence deaths annually.
UCDP: Uppsala Conflict Data Program https://ucdp.uu.se/ Uppsala Conflict Data Program (UCDP): Tracks one-sided violence (organized actors attacking unarmed civilians) UCDP definition: Conflicts causing at least 25 battle-related deaths in calendar year 2023 total organized violence: 154,000 deaths; Non-state conflicts: 20,900 deaths UCDP collects data on state-based conflicts, non-state conflicts, and one-sided violence Specific "2,700 annually" figure for state violence not found in recent UCDP data; actual figures vary annually Additional sources: https://ucdp.uu.se/ | https://en.wikipedia.org/wiki/Uppsala_Conflict_Data_Program | https://ourworldindata.org/grapher/deaths-in-armed-conflicts-by-region
.
55.
Our World in Data. Terror attack deaths (8,300 annually).
Our World in Data: Terrorism https://ourworldindata.org/terrorism (2024)
2023: 8,352 deaths (22% increase from 2022, highest since 2017) 2023: 3,350 terrorist incidents (22% decrease), but 56% increase in avg deaths per attack Global Terrorism Database (GTD): 200,000+ terrorist attacks recorded (2021 version) Maintained by: National Consortium for Study of Terrorism & Responses to Terrorism (START), U. of Maryland Geographic shift: Epicenter moved from Middle East to Central Sahel (sub-Saharan Africa) - now >50% of all deaths Additional sources: https://ourworldindata.org/terrorism | https://reliefweb.int/report/world/global-terrorism-index-2024 | https://www.start.umd.edu/gtd/ | https://ourworldindata.org/grapher/fatalities-from-terrorism
.
56.
Institute for Health Metrics and Evaluation (IHME). IHME global burden of disease 2021 (2.88B DALYs, 1.13B YLD).
Institute for Health Metrics and Evaluation (IHME) https://vizhub.healthdata.org/gbd-results/ (2024)
In 2021, global DALYs totaled approximately 2.88 billion, comprising 1.75 billion Years of Life Lost (YLL) and 1.13 billion Years Lived with Disability (YLD). This represents a 13% increase from 2019 (2.55B DALYs), largely attributable to COVID-19 deaths and aging populations. YLD accounts for approximately 39% of total DALYs, reflecting the substantial burden of non-fatal chronic conditions. Additional sources: https://vizhub.healthdata.org/gbd-results/ | https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(24)00757-8/fulltext | https://www.healthdata.org/research-analysis/about-gbd
.
57.
Costs of War Project, Brown University Watson Institute. Environmental cost of war ($100B annually).
Brown Watson Costs of War: Environmental Cost https://watson.brown.edu/costsofwar/costs/social/environment War on Terror emissions: 1.2B metric tons GHG (equivalent to 257M cars/year) Military: 5.5% of global GHG emissions (2X aviation + shipping combined) US DoD: World’s single largest institutional oil consumer, 47th largest emitter if nation Cleanup costs: $500B+ for military contaminated sites Gaza war environmental damage: $56.4B; landmine clearance: $34.6B expected Climate finance gap: Rich nations spend 30X more on military than climate finance Note: Military activities cause massive environmental damage through GHG emissions, toxic contamination, and long-term cleanup costs far exceeding current climate finance commitments Additional sources: https://watson.brown.edu/costsofwar/costs/social/environment | https://earth.org/environmental-costs-of-wars/ | https://transformdefence.org/transformdefence/stats/
.
58.
ScienceDaily. Medical research lives saved annually (4.2 million).
ScienceDaily: Physical Activity Prevents 4M Deaths https://www.sciencedaily.com/releases/2020/06/200617194510.htm (2020)
Physical activity: 3.9M early deaths averted annually worldwide (15% lower premature deaths than without) COVID vaccines (2020-2024): 2.533M deaths averted, 14.8M life-years preserved; first year alone: 14.4M deaths prevented Cardiovascular prevention: 3 interventions could delay 94.3M deaths over 25 years (antihypertensives alone: 39.4M) Pandemic research response: Millions of deaths averted through rapid vaccine/drug development Additional sources: https://www.sciencedaily.com/releases/2020/06/200617194510.htm | https://pmc.ncbi.nlm.nih.gov/articles/PMC9537923/ | https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.118.038160 | https://pmc.ncbi.nlm.nih.gov/articles/PMC9464102/
.
59.
SIPRI. 36:1 disparity ratio of spending on weapons over cures.
SIPRI: Military Spending https://www.sipri.org/commentary/blog/2016/opportunity-cost-world-military-spending (2016)
Global military spending: $2.7 trillion (2024, SIPRI) Global government medical research: $68 billion (2024) Actual ratio: 39.7:1 in favor of weapons over medical research Military R&D alone: $85B (2004 data, 10% of global R&D) Military spending increases crowd out health: 1% ↑ military = 0.62% ↓ health spending Note: Ratio actually worse than 36:1. Each 1% increase in military spending reduces health spending by 0.62%, with effect more intense in poorer countries (0.962% reduction) Additional sources: https://www.sipri.org/commentary/blog/2016/opportunity-cost-world-military-spending | https://pmc.ncbi.nlm.nih.gov/articles/PMC9174441/ | https://www.congress.gov/crs-product/R45403
.
60.
Think by Numbers. Lost human capital due to war ($270B annually).
Think by Numbers https://thinkbynumbers.org/military/war/the-economic-case-for-peace-a-comprehensive-financial-analysis/ (2021)
Lost human capital from war: $300B annually (economic impact of losing skilled/productive individuals to conflict) Broader conflict/violence cost: $14T/year globally 1.4M violent deaths/year; conflict holds back economic development, causes instability, widens inequality, erodes human capital 2002: 48.4M DALYs lost from 1.6M violence deaths = $151B economic value (2000 USD) Economic toll includes: commodity prices, inflation, supply chain disruption, declining output, lost human capital Additional sources: https://thinkbynumbers.org/military/war/the-economic-case-for-peace-a-comprehensive-financial-analysis/ | https://www.weforum.org/stories/2021/02/war-violence-costs-each-human-5-a-day/ | https://pubmed.ncbi.nlm.nih.gov/19115548/
.
61.
PubMed. Psychological impact of war cost ($100B annually).
PubMed: Economic Burden of PTSD https://pubmed.ncbi.nlm.nih.gov/35485933/ PTSD economic burden (2018 U.S.): $232.2B total ($189.5B civilian, $42.7B military) Civilian costs driven by: Direct healthcare ($66B), unemployment ($42.7B) Military costs driven by: Disability ($17.8B), direct healthcare ($10.1B) Exceeds costs of other mental health conditions (anxiety, depression) War-exposed populations: 2-3X higher rates of anxiety, depression, PTSD; women and children most vulnerable Note: Actual burden $232B, significantly higher than "$100B" claimed Additional sources: https://pubmed.ncbi.nlm.nih.gov/35485933/ | https://news.va.gov/103611/study-national-economic-burden-of-ptsd-staggering/ | https://pmc.ncbi.nlm.nih.gov/articles/PMC9957523/
.
62.
CGDev. UNHCR average refugee support cost.
CGDev https://www.cgdev.org/blog/costs-hosting-refugees-oecd-countries-and-why-uk-outlier (2024)
The average cost of supporting a refugee is $1,384 per year. This represents total host country costs (housing, healthcare, education, security). OECD countries average $6,100 per refugee (mean 2022-2023), with developing countries spending $700-1,000. Global weighted average of $1,384 is reasonable given that 75-85% of refugees are in low/middle-income countries. Additional sources: https://www.cgdev.org/blog/costs-hosting-refugees-oecd-countries-and-why-uk-outlier | https://www.unhcr.org/sites/default/files/2024-11/UNHCR-WB-global-cost-of-refugee-inclusion-in-host-country-health-systems.pdf
.
63.
World Bank. World bank trade disruption cost from conflict.
World Bank https://www.worldbank.org/en/topic/trade/publication/trading-away-from-conflict Estimated $616B annual cost from conflict-related trade disruption. World Bank research shows civil war costs an average developing country 30 years of GDP growth, with 20 years needed for trade to return to pre-war levels. Trade disputes analysis shows tariff escalation could reduce global exports by up to $674 billion. Additional sources: https://www.worldbank.org/en/topic/trade/publication/trading-away-from-conflict | https://www.nber.org/papers/w11565 | http://blogs.worldbank.org/en/trade/impacts-global-trade-and-income-current-trade-disputes
.
64.
VA. Veteran healthcare cost projections.
VA https://department.va.gov/wp-content/uploads/2025/06/2026-Budget-in-Brief.pdf (2026)
VA budget: $441.3B requested for FY 2026 (10% increase). Disability compensation: $165.6B in FY 2024 for 6.7M veterans. PACT Act projected to increase spending by $300B between 2022-2031. Costs under Toxic Exposures Fund: $20B (2024), $30.4B (2025), $52.6B (2026). Additional sources: https://department.va.gov/wp-content/uploads/2025/06/2026-Budget-in-Brief.pdf | https://www.cbo.gov/publication/45615 | https://www.legion.org/information-center/news/veterans-healthcare/2025/june/va-budget-tops-400-billion-for-2025-from-higher-spending-on-mandated-benefits-medical-care
.
67.
Cybersecurity Ventures. Cybercrime economy projected to reach $10.5 trillion.
Cybersecurity Ventures: $10.5T Cybercrime https://cybersecurityventures.com/hackerpocalypse-cybercrime-report-2016/ (2016)
Global cybercrime costs: $3T (2015) → $6T (2021) → $10.5T (2025 projected) 15% annual growth rate If measured as country, would be 3rd largest economy after US and China Greatest transfer of economic wealth in history Note: More profitable than global trade of all major illegal drugs combined. Includes data theft, productivity loss, IP theft, fraud Additional sources: <https://cybersecurityventures.com/hackerpocalypse-cybercrime-report-2016/> | https://www.boisestate.edu/cybersecurity/2022/06/16/cybercrime-to-cost-the-world-10-5-trillion-annually-by-2025/
.
69.
Bolt, J. & Zanden, J. L. van.
Maddison project database 2020. (2020)
Historical GDP per capita estimates from year 1 to present. Global GDP per capita in 1900: approximately 1,260 in 1990 international dollars (roughly 3,150 in 2024 USD after PPP and inflation adjustment). Standard reference for long-run comparative economic history.
70.
Applied Clinical Trials. Global government spending on interventional clinical trials: $3-6 billion/year.
Applied Clinical Trials https://www.appliedclinicaltrialsonline.com/view/sizing-clinical-research-market Estimated range based on NIH ( $0.8-5.6B), NIHR ($1.6B total budget), and EU funding ( $1.3B/year). Roughly 5-10% of global market. Additional sources: https://www.appliedclinicaltrialsonline.com/view/sizing-clinical-research-market | https://www.thelancet.com/journals/langlo/article/PIIS2214-109X(20)30357-0/fulltext
.
75.
United Nations Department of Economic and Social Affairs, Population Division.
World population prospects 2024: Summary of results. (2024)
The 2024 Revision of the World Population Prospects provides population estimates and projections for 237 countries or areas. Global median age approximately 30.5 years in 2024, reflecting population-weighted average across all regions.
78.
Estimated from major foundation budgets and activities. Nonprofit clinical trial funding estimate.
Nonprofit foundations spend an estimated $2-5 billion annually on clinical trials globally, representing approximately 2-5% of total clinical trial spending.
79.
ICAN. Global nuclear weapon maintenance cost: $100 billion/year.
ICAN: Global Spending $100B 2024 https://www.icanw.org/global_spending_on_nuclear_weapons_topped_100_billion_in_2024 (2024)
2024: >$100 billion ($190,151/minute) - 11% increase ($9.9B) from 2023 Nine nuclear-armed states: China, France, India, Israel, N. Korea, Pakistan, Russia, UK, US US: $56.8B (more than all other 8 states combined); China: $12.5B; UK: $10B (+26% YoY, biggest increase) Historical trend: $72.9B (2019) → $82.4B (2021) → >$100B (2024) Private sector contracts: $463B ongoing; $42.5B earned from contracts in 2024 alone Note: $100B/year figure accurate for 2024. Rapid growth from $73B (2019). US spends more than rest of world combined on nuclear weapons Additional sources: https://www.icanw.org/global_spending_on_nuclear_weapons_topped_100_billion_in_2024 | https://www.icanw.org/the_cost_of_nuclear_weapons
.
80.
Industry reports: IQVIA. Global pharmaceutical r&d spending.
Total global pharmaceutical R&D spending is approximately $300 billion annually. Clinical trials represent 15-20% of this total ($45-60B), with the remainder going to drug discovery, preclinical research, regulatory affairs, and manufacturing development.
81.
UN. Global population reaches 8 billion.
UN: World Population 8 Billion Nov 15 2022 https://www.un.org/en/desa/world-population-reach-8-billion-15-november-2022 (2022)
Milestone: November 15, 2022 (UN World Population Prospects 2022) Day of Eight Billion" designated by UN Added 1 billion people in just 11 years (2011-2022) Growth rate: Slowest since 1950; fell under 1% in 2020 Future: 15 years to reach 9B (2037); projected peak 10.4B in 2080s Projections: 8.5B (2030), 9.7B (2050), 10.4B (2080-2100 plateau) Note: Milestone reached Nov 2022. Population growth slowing; will take longer to add next billion (15 years vs 11 years) Additional sources: https://www.un.org/en/desa/world-population-reach-8-billion-15-november-2022 | https://www.un.org/en/dayof8billion | https://en.wikipedia.org/wiki/Day_of_Eight_Billion
.
82.
Harvard Kennedy School. 3.5% participation tipping point.
Harvard Kennedy School https://www.hks.harvard.edu/centers/carr/publications/35-rule-how-small-minority-can-change-world (2020)
The research found that nonviolent campaigns were twice as likely to succeed as violent ones, and once 3.5% of the population were involved, they were always successful. Chenoweth and Maria Stephan studied the success rates of civil resistance efforts from 1900 to 2006, finding that nonviolent movements attracted, on average, four times as many participants as violent movements and were more likely to succeed. Key finding: Every campaign that mobilized at least 3.5% of the population in sustained protest was successful (in their 1900-2006 dataset) Note: The 3.5% figure is a descriptive statistic from historical analysis, not a guaranteed threshold. One exception (Bahrain 2011-2014 with 6%+ participation) has been identified. The rule applies to regime change, not policy change in democracies. Additional sources: https://www.hks.harvard.edu/centers/carr/publications/35-rule-how-small-minority-can-change-world | https://www.hks.harvard.edu/sites/default/files/2024-05/Erica%20Chenoweth_2020-005.pdf | https://www.bbc.com/future/article/20190513-it-only-takes-35-of-people-to-change-the-world | https://en.wikipedia.org/wiki/3.5%25_rule
.
83.
International IDEA.
International IDEA voter turnout database world export. (2026)
Best current register-based estimate of global registered voters. Sum of the latest available country-level Registration counts in International IDEA’s world export on 2026-04-22 = 4,128,142,495 registered voters across 199 countries and political entities. Methodology notes that Registration is the number of names on the voters’ register as reported by electoral management bodies, and comparability is imperfect because voter rolls and registration systems differ across countries. Additional sources: https://www.idea.int/data-tools/data/voter-turnout-database | https://www.idea.int/data-tools/export?type=region_only&themeId=293&world=all&loc=home
.
85.
Federation of American Scientists. World nuclear forces.
Federation of American Scientists https://fas.org/issues/nuclear-weapons/status-world-nuclear-forces/ (2024)
As of early 2025, we estimate that the world’s nine nuclear-armed states possess a combined total of approximately 12,241 nuclear warheads. Additional sources: https://fas.org/issues/nuclear-weapons/status-world-nuclear-forces/
.
86.
OpenSecrets.
Top lobbying industries 2025. (2025)
Sector ranks and per-company federal lobbying spending for 2025. Combined market capitalization of the top-5 publicly traded US lobbying spenders in each government-controlling sector: pharmaceuticals $1,794.7B; technology $13,279.5B; insurance $385.6B; oil and gas $1,246.9B; four-sector total approx $16.71T. Caveats: Meta (Zuckerberg holds 60.8% of voting power) and Alphabet (Page and Brin hold 52.3%) cannot be majority-acquired; Ellison owns 40.6% of Oracle; the largest insurance lobbyists are mutuals with no public shares; trade associations (PhRMA, AHIP, SIFMA, API) are not acquirable. Additional sources: https://stockanalysis.com/stocks/
.
87.
NHGRI. Human genome project and CRISPR discovery.
NHGRI https://www.genome.gov/11006929/2003-release-international-consortium-completes-hgp (2003)
Your DNA is 3 billion base pairs Read the entire code (Human Genome Project, completed 2003) Learned to edit it (CRISPR, discovered 2012) Additional sources: https://www.genome.gov/11006929/2003-release-international-consortium-completes-hgp | https://www.nobelprize.org/prizes/chemistry/2020/press-release/
.
88.
PMC. Only 12% of human interactome targeted.
PMC https://pmc.ncbi.nlm.nih.gov/articles/PMC10749231/ (2023)
Mapping 350,000+ clinical trials showed that only 12% of the human interactome has ever been targeted by drugs. Additional sources: https://pmc.ncbi.nlm.nih.gov/articles/PMC10749231/
.
89.
WHO. ICD-10 code count ( 14,000).
WHO https://icd.who.int/browse10/2019/en (2019)
The ICD-10 classification contains approximately 14,000 codes for diseases, signs and symptoms. Additional sources: https://icd.who.int/browse10/2019/en
.
91.
McFarland, M. J., Hauer, M. E. & Reuben, A.
Half of US population exposed to adverse lead levels in early childhood.
Proceedings of the National Academy of Sciences 119, e2118631119 (2022)
Leaded gasoline, used in the US from 1923 until its on-road ban in 1996, exposed more than half of the 2015 US population to adverse blood-lead levels in early childhood. The authors estimate childhood lead exposure cost the population a cumulative 824 million IQ points, an average of 2.6 points per person, rising to 5.9 points for the most-exposed 1966-1970 birth cohort.
92.
Wikipedia. Longevity escape velocity (LEV) - maximum human life extension potential.
Wikipedia: Longevity Escape Velocity https://en.wikipedia.org/wiki/Longevity_escape_velocity Longevity escape velocity: Hypothetical point where medical advances extend life expectancy faster than time passes Term coined by Aubrey de Grey (biogerontologist) in 2004 paper; concept from David Gobel (Methuselah Foundation) Current progress: Science adds 3 months to lifespan per year; LEV requires adding >1 year per year Sinclair (Harvard): "There is no biological upper limit to age" - first person to live to 150 may already be born De Grey: 50% chance of reaching LEV by mid-to-late 2030s; SENS approach = damage repair rather than slowing damage Kurzweil (2024): LEV by 2029-2035, AI will simulate biological processes to accelerate solutions George Church: LEV "in a decade or two" via age-reversal clinical trials Natural lifespan cap: 120-150 years (Jeanne Calment record: 122); engineering approach could bypass via damage repair Key mechanisms: Epigenetic reprogramming, senolytic drugs, stem cell therapy, gene therapy, AI-driven drug discovery Current record: Jeanne Calment (122 years, 164 days) - record unbroken since 1997 Note: LEV is theoretical but increasingly plausible given demonstrated age reversal in mice (109% lifespan extension) and human cells (30-year epigenetic age reversal) Additional sources: https://en.wikipedia.org/wiki/Longevity_escape_velocity | https://pmc.ncbi.nlm.nih.gov/articles/PMC423155/ | https://www.popularmechanics.com/science/a36712084/can-science-cure-death-longevity/ | https://www.diamandis.com/blog/longevity-escape-velocity
.
95.
OpenSecrets. Lobbyist statistics for washington d.c.
OpenSecrets: Lobbying in US https://en.wikipedia.org/wiki/Lobbying_in_the_United_States Registered lobbyists: Over 12,000 (some estimates); 12,281 registered (2013) Former government employees as lobbyists: 2,200+ former federal employees (1998-2004), including 273 former White House staffers, 250 former Congress members & agency heads Congressional revolving door: 43% (86 of 198) lawmakers who left 1998-2004 became lobbyists; currently 59% leaving to private sector work for lobbying/consulting firms/trade groups Executive branch: 8% were registered lobbyists at some point before/after government service Additional sources: https://en.wikipedia.org/wiki/Lobbying_in_the_United_States | https://www.opensecrets.org/revolving-door | https://www.citizen.org/article/revolving-congress/ | https://www.propublica.org/article/we-found-a-staggering-281-lobbyists-whove-worked-in-the-trump-administration
.
96.
MDPI Vaccines. Measles vaccination ROI.
MDPI Vaccines https://www.mdpi.com/2076-393X/12/11/1210 (2024)
Single measles vaccination: 167:1 benefit-cost ratio. MMR (measles-mumps-rubella) vaccination: 14:1 ROI. Historical US elimination efforts (1966-1974): benefit-cost ratio of 10.3:1 with net benefits exceeding USD 1.1 billion (1972 dollars, or USD 8.0 billion in 2023 dollars). 2-dose MMR programs show direct benefit/cost ratio of 14.2 with net savings of $5.3 billion, and 26.0 from societal perspectives with net savings of $11.6 billion. Additional sources: https://www.mdpi.com/2076-393X/12/11/1210 | https://www.tandfonline.com/doi/full/10.1080/14760584.2024.2367451
.
100.
U.S. Government Accountability Office.
Electronic Health Records: First Year of CMS’s Incentive Programs Shows Opportunities to Improve Processes to Verify Providers Met Requirements.
https://www.gao.gov/products/gao-12-481 (2012).
106.
Calculated from Orphanet Journal of Rare Diseases (2024). Diseases getting first effective treatment each year.
Calculated from Orphanet Journal of Rare Diseases (2024) https://ojrd.biomedcentral.com/articles/10.1186/s13023-024-03398-1 (2024)
Under the current system, approximately 10-15 diseases per year receive their FIRST effective treatment. Calculation: 5% of 7,000 rare diseases ( 350) have FDA-approved treatment, accumulated over 40 years of the Orphan Drug Act = 9 rare diseases/year. Adding 5-10 non-rare diseases that get first treatments yields 10-20 total. FDA approves 50 drugs/year, but many are for diseases that already have treatments (me-too drugs, second-line therapies). Only 15 represent truly FIRST treatments for previously untreatable conditions.
107.
NIH. NIH budget (FY 2025).
NIH https://www.nih.gov/about-nih/organization/budget (2024)
The budget total of $47.7 billion also includes $1.412 billion derived from PHS Evaluation financing... Additional sources: https://www.nih.gov/about-nih/organization/budget | https://officeofbudget.od.nih.gov/
.
108.
Bentley et al. NIH spending on clinical trials: 3.3%.
Bentley et al. https://pmc.ncbi.nlm.nih.gov/articles/PMC10349341/ (2023)
NIH spent $8.1 billion on clinical trials for approved drugs (2010-2019), representing 3.3% of relevant NIH spending. Additional sources: https://pmc.ncbi.nlm.nih.gov/articles/PMC10349341/ | https://catalyst.harvard.edu/news/article/nih-spent-8-1b-for-phased-clinical-trials-of-drugs-approved-2010-19-10-of-reported-industry-spending/
.
109.
PMC. Standard medical research ROI ($20k-$100k/QALY).
PMC: Cost-effectiveness Thresholds Used by Study Authors https://pmc.ncbi.nlm.nih.gov/articles/PMC10114019/ (1990)
Typical cost-effectiveness thresholds for medical interventions in rich countries range from $50,000 to $150,000 per QALY. The Institute for Clinical and Economic Review (ICER) uses a $100,000-$150,000/QALY threshold for value-based pricing. Between 1990-2021, authors increasingly cited $100,000 (47% by 2020-21) or $150,000 (24% by 2020-21) per QALY as benchmarks for cost-effectiveness. Additional sources: https://pmc.ncbi.nlm.nih.gov/articles/PMC10114019/ | https://icer.org/our-approach/methods-process/cost-effectiveness-the-qaly-and-the-evlyg/
.
110.
Xia et al., Nature Food. Nuclear winter famine.
Xia et al. https://www.nature.com/articles/s43016-022-00573-0 (2022)
We estimate that a nuclear war between the United States and Russia would produce 150 Tg of soot and lead to 5 billion people dying at the end of year 2. Additional sources: https://www.nature.com/articles/s43016-022-00573-0
.
111.
Manhattan Institute. RECOVERY trial 82× cost reduction.
Manhattan Institute: Slow Costly Trials https://manhattan.institute/article/slow-costly-clinical-trials-drag-down-biomedical-breakthroughs RECOVERY trial: $500 per patient ($20M for 48,000 patients = $417/patient) Typical clinical trial: $41,000 median per-patient cost Cost reduction: 80-82× cheaper ($41,000 ÷ $500 ≈ 82×) Efficiency: $50 per patient per answer (10 therapeutics tested, 4 effective) Dexamethasone estimated to save >630,000 lives Additional sources: https://manhattan.institute/article/slow-costly-clinical-trials-drag-down-biomedical-breakthroughs | https://pmc.ncbi.nlm.nih.gov/articles/PMC9293394/
.
112.
Trials. Patient willingness to participate in clinical trials.
Trials: Patients’ Willingness Survey https://trialsjournal.biomedcentral.com/articles/10.1186/s13063-015-1105-3 Recent surveys: 49-51% willingness (2020-2022) - dramatic drop from 85% (2019) during COVID-19 pandemic Cancer patients when approached: 88% consented to trials (Royal Marsden Hospital) Study type variation: 44.8% willing for drug trial, 76.2% for diagnostic study Top motivation: "Learning more about my health/medical condition" (67.4%) Top barrier: "Worry about experiencing side effects" (52.6%) Additional sources: https://trialsjournal.biomedcentral.com/articles/10.1186/s13063-015-1105-3 | https://www.appliedclinicaltrialsonline.com/view/industry-forced-to-rethink-patient-participation-in-trials | https://pmc.ncbi.nlm.nih.gov/articles/PMC7183682/
.
113.
The Commune. Pentagon audit failures ($2.46T unaccounted).
The Commune https://thecommunemag.com/the-pentagon-misplaced-2-46-trillion-an-in-depth-look-at-the-financial-audit-failures (2024)
In the most recent audit, the Department of Defense (DoD) could not account for approximately 60% of its \(4.1 trillion in assets, amounting to\)2.46 trillion unaccounted for. Alternative title: Pentagon unsupported accounting adjustments (\(6.5T, single year, US Army) In 2015, the Department of Defense's Inspector General reported that the Army could not adequately support\)6.5 trillion in year-end adjustments, indicating severe accounting discrepancies. Additional sources: https://thecommunemag.com/the-pentagon-misplaced-2-46-trillion-an-in-depth-look-at-the-financial-audit-failures | https://accmag.com/audit-pentagon-cannot-account-for-6-5-trillion-dollars-is-taxpayer-money/
.
114.
Tufts CSDD. Cost of drug development.
Various estimates suggest $1.0 - $2.5 billion to bring a new drug from discovery through FDA approval, spread across 10 years. Tufts Center for the Study of Drug Development often cited for $1.0 - $2.6 billion/drug. Industry reports (IQVIA, Deloitte) also highlight $2+ billion figures.
115.
Value in Health. Average lifetime revenue per successful drug.
Value in Health: Sales Revenues for New Therapeutic Agents https://www.sciencedirect.com/science/article/pii/S1098301524027542 Study of 361 FDA-approved drugs from 1995-2014 (median follow-up 13.2 years): Mean lifetime revenue: $15.2 billion per drug Median lifetime revenue: $6.7 billion per drug Revenue after 5 years: $3.2 billion (mean) Revenue after 10 years: $9.5 billion (mean) Revenue after 15 years: $19.2 billion (mean) Distribution highly skewed: top 25 drugs (7%) accounted for 38% of total revenue ($2.1T of $5.5T) Additional sources: https://www.sciencedirect.com/science/article/pii/S1098301524027542
.
116.
Lichtenberg, F. R.
How many life-years have new drugs saved? A three-way fixed-effects analysis of 66 diseases in 27 countries, 2000-2013.
International Health 11, 403–416 (2019)
Using 3-way fixed-effects methodology (disease-country-year) across 66 diseases in 22 countries, this study estimates that drugs launched after 1981 saved 148.7 million life-years in 2013 alone. The regression coefficients for drug launches 0-11 years prior (beta=-0.031, SE=0.008) and 12+ years prior (beta=-0.057, SE=0.013) on years of life lost are highly significant (p<0.0001). Confidence interval for life-years saved: 79.4M-239.8M (95 percent CI) based on propagated standard errors from Table 2.
117.
Deloitte. Pharmaceutical r&d return on investment (ROI).
Deloitte: Measuring Pharmaceutical Innovation 2025 https://www.deloitte.com/ch/en/Industries/life-sciences-health-care/research/measuring-return-from-pharmaceutical-innovation.html (2025)
Deloitte’s annual study of top 20 pharma companies by R&D spend (2010-2024): 2024 ROI: 5.9% (second year of growth after decade of decline) 2023 ROI: 4.3% (estimated from trend) 2022 ROI: 1.2% (historic low since study began, 13-year low) 2021 ROI: 6.8% (record high, inflated by COVID-19 vaccines/treatments) Long-term trend: Declining for over a decade before 2023 recovery Average R&D cost per asset: $2.3B (2022), $2.23B (2024) These returns (1.2-5.9% range) fall far below typical corporate ROI targets (15-20%) Additional sources: https://www.deloitte.com/ch/en/Industries/life-sciences-health-care/research/measuring-return-from-pharmaceutical-innovation.html | https://www.prnewswire.com/news-releases/deloittes-13th-annual-pharmaceutical-innovation-report-pharma-rd-return-on-investment-falls-in-post-pandemic-market-301738807.html | https://hitconsultant.net/2023/02/16/pharma-rd-roi-falls-to-lowest-level-in-13-years/
.
118.
Nature Reviews Drug Discovery. Drug trial success rate from phase i to approval.
Nature Reviews Drug Discovery: Clinical Success Rates https://www.nature.com/articles/nrd.2016.136 (2016)
Overall Phase I to approval: 10-12.8% (conventional wisdom 10%, studies show 12.8%) Recent decline: Average LOA now 6.7% for Phase I (2014-2023 data) Leading pharma companies: 14.3% average LOA (range 8-23%) Varies by therapeutic area: Oncology 3.4%, CNS/cardiovascular lowest at Phase III Phase-specific success: Phase I 47-54%, Phase II 28-34%, Phase III 55-70% Note: 12% figure accurate for historical average. Recent data shows decline to 6.7%, with Phase II as primary attrition point (28% success) Additional sources: https://www.nature.com/articles/nrd.2016.136 | https://pmc.ncbi.nlm.nih.gov/articles/PMC6409418/ | https://academic.oup.com/biostatistics/article/20/2/273/4817524
.
119.
SofproMed. Phase 3 cost per trial range.
SofproMed https://www.sofpromed.com/how-much-does-a-clinical-trial-cost Phase 3 clinical trials cost between $20 million and $282 million per trial, with significant variation by therapeutic area and trial complexity. Additional sources: https://www.sofpromed.com/how-much-does-a-clinical-trial-cost | https://www.cbo.gov/publication/57126
.
120.
Ramsberg, J. & Platt, R. Pragmatic trial cost per patient (median $97).
Learning Health Systems https://pmc.ncbi.nlm.nih.gov/articles/PMC6508852/ (2018)
Meta-analysis of 108 embedded pragmatic clinical trials (2006-2016). The median cost per patient was $97 (IQR $19–$478), based on 2015 dollars. 25% of trials cost <$19/patient; 10 trials exceeded $1,000/patient. U.S. studies median $187 vs non-U.S. median $27. Additional sources: https://pmc.ncbi.nlm.nih.gov/articles/PMC6508852/
.
121.
WHO. Polio vaccination ROI.
WHO https://www.who.int/news-room/feature-stories/detail/sustaining-polio-investments-offers-a-high-return (2019)
For every dollar spent, the return on investment is nearly US$ 39." Total investment cost of US$ 7.5 billion generates projected economic and social benefits of US$ 289.2 billion from sustaining polio assets and integrating them into expanded immunization, surveillance and emergency response programmes across 8 priority countries (Afghanistan, Iraq, Libya, Pakistan, Somalia, Sudan, Syria, Yemen). Additional sources: https://www.who.int/news-room/feature-stories/detail/sustaining-polio-investments-offers-a-high-return
.
122.
ICRC. International campaign to ban landmines (ICBL) - ottawa treaty (1997).
ICRC https://www.icrc.org/en/doc/resources/documents/article/other/57jpjn.htm (1997)
ICBL: Founded 1992 by 6 NGOs (Handicap International, Human Rights Watch, Medico International, Mines Advisory Group, Physicians for Human Rights, Vietnam Veterans of America Foundation) Started with ONE staff member: Jody Williams as founding coordinator Grew to 1,000+ organizations in 60 countries by 1997 Ottawa Process: 14 months (October 1996 - December 1997) Convention signed by 122 states on December 3, 1997; entered into force March 1, 1999 Achievement: Nobel Peace Prize 1997 (shared by ICBL and Jody Williams) Government funding context: Canada established $100M CAD Canadian Landmine Fund over 10 years (1997); International donors provided $169M in 1997 for mine action (up from $100M in 1996) Additional sources: https://www.icrc.org/en/doc/resources/documents/article/other/57jpjn.htm | https://en.wikipedia.org/wiki/International_Campaign_to_Ban_Landmines | https://www.nobelprize.org/prizes/peace/1997/summary/ | https://un.org/press/en/1999/19990520.MINES.BRF.html | https://www.the-monitor.org/en-gb/reports/2003/landmine-monitor-2003/mine-action-funding.aspx
.
123.
OpenSecrets.
Revolving door: Former members of congress. (2024)
388 former members of Congress are registered as lobbyists. Nearly 5,400 former congressional staffers have left Capitol Hill to become federal lobbyists in the past 10 years. Additional sources: https://www.opensecrets.org/revolving-door
.
124.
Kinch, M. S. & Griesenauer, R. H.
Lost medicines: A longer view of the pharmaceutical industry with the potential to reinvigorate discovery.
Drug Discovery Today 24, 875–880 (2019)
Research identified 1,600+ medicines available in 1962. The 1950s represented industry high-water mark with >30 new products in five of ten years; this rate would not be replicated until late 1990s. More than half (880) of these medicines were lost following implementation of Kefauver-Harris Amendment. The peak of 1962 would not be seen again until early 21st century. By 2016 number of organizations actively involved in R&D at level not seen since 1914.
125.
Baily, M. N. Pre-1962 drug development costs (baily 1972).
Baily (1972) https://samizdathealth.org/wp-content/uploads/2020/12/hlthaff.1.2.6.pdf (1972)
Pre-1962: Average cost per new chemical entity (NCE) was $6.5 million (1980 dollars) Inflation-adjusted to 2024 dollars: $6.5M (1980) ≈ $22.5M (2024), using CPI multiplier of 3.46× Real cost increase (inflation-adjusted): $22.5M (pre-1962) → $2,600M (2024) = 116× increase Note: This represents the most comprehensive academic estimate of pre-1962 drug development costs based on empirical industry data Additional sources: https://samizdathealth.org/wp-content/uploads/2020/12/hlthaff.1.2.6.pdf
.
126.
Think by Numbers. Pre-1962 physician-led clinical trials.
Think by Numbers: How Many Lives Does FDA Save? https://thinkbynumbers.org/health/how-many-net-lives-does-the-fda-save/ (1966)
Pre-1962: Physicians could report real-world evidence directly 1962 Drug Amendments replaced "premarket notification" with "premarket approval", requiring extensive efficacy testing Impact: New regulatory clampdown reduced new treatment production by 70%; lifespan growth declined from 4 years/decade to 2 years/decade Drug Efficacy Study Implementation (DESI): NAS/NRC evaluated 3,400+ drugs approved 1938-1962 for safety only; reviewed >3,000 products, >16,000 therapeutic claims FDA has had authority to accept real-world evidence since 1962, clarified by 21st Century Cures Act (2016) Note: Specific "144,000 physicians" figure not verified in sources Additional sources: https://thinkbynumbers.org/health/how-many-net-lives-does-the-fda-save/ | https://www.fda.gov/drugs/enforcement-activities-fda/drug-efficacy-study-implementation-desi | http://www.nasonline.org/about-nas/history/archives/collections/des-1966-1969-1.html
.
127.
GAO. 95% of diseases have 0 FDA-approved treatments.
GAO https://www.gao.gov/products/gao-25-106774 (2025)
95% of diseases have no treatment Additional sources: https://www.gao.gov/products/gao-25-106774 | https://globalgenes.org/rare-disease-facts/
.
129.
NHS England; Águas et al. RECOVERY trial global lives saved ( 1 million).
NHS England: 1 Million Lives Saved https://www.england.nhs.uk/2021/03/covid-treatment-developed-in-the-nhs-saves-a-million-lives/ (2021)
Dexamethasone saved 1 million lives worldwide (NHS England estimate, March 2021, 9 months after discovery). UK alone: 22,000 lives saved. Methodology: Águas et al. Nature Communications 2021 estimated 650,000 lives (range: 240,000-1,400,000) for July-December 2020 alone, based on RECOVERY trial mortality reductions (36% for ventilated, 18% for oxygen-only patients) applied to global COVID hospitalizations. June 2020 announcement: Dexamethasone reduced deaths by up to 1/3 (ventilated patients), 1/5 (oxygen patients). Impact immediate: Adopted into standard care globally within hours of announcement. Additional sources: https://www.england.nhs.uk/2021/03/covid-treatment-developed-in-the-nhs-saves-a-million-lives/ | https://www.nature.com/articles/s41467-021-21134-2 | https://pharmaceutical-journal.com/article/news/steroid-has-saved-the-lives-of-one-million-covid-19-patients-worldwide-figures-show | https://www.recoverytrial.net/news/recovery-trial-celebrates-two-year-anniversary-of-life-saving-dexamethasone-result
.
132.
National September 11 Memorial & Museum.
September 11 attack facts. (2024)
2,977 people were killed in the September 11, 2001 attacks: 2,753 at the World Trade Center, 184 at the Pentagon, and 40 passengers and crew on United Flight 93 in Shanksville, Pennsylvania.
133.
World Bank. World bank singapore economic data.
World Bank https://data.worldbank.org/country/singapore (2024)
Singapore GDP per capita (2023): $82,000 - among highest in the world Government spending: 15% of GDP (vs US 38%) Life expectancy: 84.1 years (vs US 77.5 years) Singapore demonstrates that low government spending can coexist with excellent outcomes Additional sources: https://data.worldbank.org/country/singapore
.
134.
International Monetary Fund.
IMF singapore government spending data. (2024)
Singapore government spending is approximately 15% of GDP This is 23 percentage points lower than the United States (38%) Despite lower spending, Singapore achieves excellent outcomes: - Life expectancy: 84.1 years (vs US 77.5) - Low crime, world-class infrastructure, AAA credit rating Additional sources: https://www.imf.org/en/Countries/SGP
.
135.
World Health Organization.
WHO life expectancy data by country. (2024)
Life expectancy at birth varies significantly among developed nations: Switzerland: 84.0 years (2023) Singapore: 84.1 years (2023) Japan: 84.3 years (2023) United States: 77.5 years (2023) - 6.5 years below Switzerland, Singapore Global average: 73 years Note: US spends more per capita on healthcare than any other nation, yet achieves lower life expectancy Additional sources: https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-life-expectancy-and-healthy-life-expectancy
.
137.
PMC. Contribution of smoking reduction to life expectancy gains.
PMC: Benefits Smoking Cessation Longevity https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1447499/ (2012)
Population-level: Up to 14% (9% men, 14% women) of total life expectancy gain since 1960 due to tobacco control efforts Individual cessation benefits: Quitting at age 35 adds 6.9-8.5 years (men), 6.1-7.7 years (women) vs continuing smokers By cessation age: Age 25-34 = 10 years gained; age 35-44 = 9 years; age 45-54 = 6 years; age 65 = 2.0 years (men), 3.7 years (women) Cessation before age 40: Reduces death risk by 90% Long-term cessation: 10+ years yields survival comparable to never smokers, averts 10 years of life lost Recent cessation: <3 years averts 5 years of life lost Additional sources: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1447499/ | https://www.cdc.gov/pcd/issues/2012/11_0295.htm | https://www.ajpmonline.org/article/S0749-3797(24)00217-4/fulltext | https://www.nejm.org/doi/full/10.1056/NEJMsa1211128
.
138.
ICER. Value per QALY (standard economic value).
ICER https://icer.org/wp-content/uploads/2024/02/Reference-Case-4.3.25.pdf (2024)
Standard economic value per QALY: $100,000–$150,000. This is the US and global standard willingness-to-pay threshold for interventions that add costs. Dominant interventions (those that save money while improving health) are favorable regardless of this threshold. Additional sources: https://icer.org/wp-content/uploads/2024/02/Reference-Case-4.3.25.pdf
.
139.
GAO. Annual cost of u.s. Sugar subsidies.
GAO: Sugar Program https://www.gao.gov/products/gao-24-106144 Consumer costs: $2.5-3.5 billion per year (GAO estimate) Net economic cost: $1 billion per year 2022: US consumers paid 2X world price for sugar Program costs $3-4 billion/year but no federal budget impact (costs passed directly to consumers via higher prices) Employment impact: 10,000-20,000 manufacturing jobs lost annually in sugar-reliant industries (confectionery, etc.) Multiple studies confirm: Sweetener Users Association ($2.9-3.5B), AEI ($2.4B consumer cost), Beghin & Elobeid ($2.9-3.5B consumer surplus) Additional sources: https://www.gao.gov/products/gao-24-106144 | https://www.heritage.org/agriculture/report/the-us-sugar-program-bad-consumers-bad-agriculture-and-bad-america | https://www.aei.org/articles/the-u-s-spends-4-billion-a-year-subsidizing-stalinist-style-domestic-sugar-production/
.
140.
World Bank. Swiss military budget as percentage of GDP.
World Bank: Military Expenditure https://data.worldbank.org/indicator/MS.MIL.XPND.GD.ZS?locations=CH 2023: 0.70272% of GDP (World Bank) 2024: CHF 5.95 billion official military spending When including militia system costs: 1% GDP (CHF 8.75B) Comparison: Near bottom in Europe; only Ireland, Malta, Moldova spend less (excluding microstates with no armies) Additional sources: https://data.worldbank.org/indicator/MS.MIL.XPND.GD.ZS?locations=CH | https://www.avenir-suisse.ch/en/blog-defence-spending-switzerland-is-in-better-shape-than-it-seems/ | https://tradingeconomics.com/switzerland/military-expenditure-percent-of-gdp-wb-data.html
.
141.
World Bank. Switzerland vs. US GDP per capita comparison.
World Bank: Switzerland GDP Per Capita https://data.worldbank.org/indicator/NY.GDP.PCAP.CD?locations=CH 2024 GDP per capita (PPP-adjusted): Switzerland $93,819 vs United States $75,492 Switzerland’s GDP per capita 24% higher than US when adjusted for purchasing power parity Nominal 2024: Switzerland $103,670 vs US $85,810 Additional sources: https://data.worldbank.org/indicator/NY.GDP.PCAP.CD?locations=CH | https://tradingeconomics.com/switzerland/gdp-per-capita-ppp | https://www.theglobaleconomy.com/USA/gdp_per_capita_ppp/
.
142.
OECD.
OECD government spending as percentage of GDP. (2024)
OECD government spending data shows significant variation among developed nations: United States: 38.0% of GDP (2023) Switzerland: 35.0% of GDP - 3 percentage points lower than US Singapore: 15.0% of GDP - 23 percentage points lower than US (per IMF data) OECD average: approximately 40% of GDP Additional sources: https://data.oecd.org/gga/general-government-spending.htm
.
143.
OECD.
OECD median household income comparison. (2024)
Median household disposable income varies significantly across OECD nations: United States: $77,500 (2023) Switzerland: $55,000 PPP-adjusted (lower nominal but comparable purchasing power) Singapore: $75,000 PPP-adjusted Additional sources: https://data.oecd.org/hha/household-disposable-income.htm
.
144.
Wikipedia. Thalidomide scandal: Worldwide cases and mortality.
Wikipedia https://en.wikipedia.org/wiki/Thalidomide_scandal The total number of embryos affected by the use of thalidomide during pregnancy is estimated at 10,000, of whom about 40% died around the time of birth. More than 10,000 children in 46 countries were born with deformities such as phocomelia. Additional sources: https://en.wikipedia.org/wiki/Thalidomide_scandal
.
145.
PLOS One. Health and quality of life of thalidomide survivors as they age.
PLOS One https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0210222 (2019)
Study of thalidomide survivors documenting ongoing disability impacts, quality of life, and long-term health outcomes. Survivors (now in their 60s) continue to experience significant disability from limb deformities, organ damage, and other effects. Additional sources: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0210222
.
147.
FDA Study via NCBI. Trial costs, FDA study.
FDA Study via NCBI https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6248200/ Overall, the 138 clinical trials had an estimated median (IQR) cost of $19.0 million ($12.2 million-$33.1 million)... The clinical trials cost a median (IQR) of $41,117 ($31,802-$82,362) per patient. Additional sources: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6248200/
.
148.
GBD 2019 Diseases and Injuries Collaborators.
Global burden of disease study 2019: Disability weights.
The Lancet 396, 1204–1222 (2020)
Disability weights for 235 health states used in Global Burden of Disease calculations. Weights range from 0 (perfect health) to 1 (death equivalent). Chronic conditions like diabetes (0.05-0.35), COPD (0.04-0.41), depression (0.15-0.66), and cardiovascular disease (0.04-0.57) show substantial variation by severity. Treatment typically reduces disability weights by 50-80 percent for manageable chronic conditions.
149.
WHO. Annual global economic burden of alzheimer’s and other dementias.
WHO: Dementia Fact Sheet https://www.who.int/news-room/fact-sheets/detail/dementia (2019)
Global cost: $1.3 trillion (2019 WHO-commissioned study) 50% from informal caregivers (family/friends, 5 hrs/day) 74% of costs in high-income countries despite 61% of patients in LMICs $818B (2010) → $1T (2018) → $1.3T (2019) - rapid growth Note: Costs increased 35% from 2010-2015 alone. Informal care represents massive hidden economic burden Additional sources: https://www.who.int/news-room/fact-sheets/detail/dementia | https://alz-journals.onlinelibrary.wiley.com/doi/10.1002/alz.12901
.
150.
JAMA Oncology. Annual global economic burden of cancer.
JAMA Oncology: Global Cost 2020-2050 https://jamanetwork.com/journals/jamaoncology/fullarticle/2801798 (2020)
2020-2050 projection: $25.2 trillion total ($840B/year average) 2010 annual cost: $1.16 trillion (direct costs only) Recent estimate: $3 trillion/year (all costs included) Top 5 cancers: lung (15.4%), colon/rectum (10.9%), breast (7.7%), liver (6.5%), leukemia (6.3%) Note: China/US account for 45% of global burden; 75% of deaths in LMICs but only 50.0% of economic cost Additional sources: https://jamanetwork.com/journals/jamaoncology/fullarticle/2801798 | https://www.nature.com/articles/d41586-023-00634-9
.
152.
Diabetes Care. Annual global economic burden of diabetes.
Diabetes Care: Global Economic Burden https://diabetesjournals.org/care/article/41/5/963/36522/Global-Economic-Burden-of-Diabetes-in-Adults 2015: $1.3 trillion (1.8% of global GDP) 2030 projections: $2.1T-2.5T depending on scenario IDF health expenditure: $760B (2019) → $845B (2045 projected) 2/3 direct medical costs ($857B), 1/3 indirect costs (lost productivity) Note: Costs growing rapidly; expected to exceed $2T by 2030 Additional sources: https://diabetesjournals.org/care/article/41/5/963/36522/Global-Economic-Burden-of-Diabetes-in-Adults | https://doi.org/10.1016/S2213-8587(17)30097-9
.
154.
World Bank, Bureau of Economic Analysis. US GDP 2024 ($28.78 trillion).
World Bank https://data.worldbank.org/indicator/NY.GDP.MKTP.CD?locations=US (2024)
US GDP reached $28.78 trillion in 2024, representing approximately 26% of global GDP. Additional sources: https://data.worldbank.org/indicator/NY.GDP.MKTP.CD?locations=US | https://www.bea.gov/news/2024/gross-domestic-product-fourth-quarter-and-year-2024-advance-estimate
.
155.
Environmental Working Group. US farm subsidy database and analysis.
Environmental Working Group https://farm.ewg.org/ (2024)
US agricultural subsidies total approximately $30 billion annually, but create much larger economic distortions. Top 10% of farms receive 78% of subsidies, benefits concentrated in commodity crops (corn, soy, wheat, cotton), environmental damage from monoculture incentivized, and overall deadweight loss estimated at $50-120 billion annually. Additional sources: https://farm.ewg.org/ | https://www.ers.usda.gov/topics/farm-economy/farm-sector-income-finances/government-payments-the-safety-net/
.
156.
Drug Policy Alliance.
The drug war by the numbers. (2021)
Since 1971, the war on drugs has cost the United States an estimated $1 trillion in enforcement. The federal drug control budget was $41 billion in 2022. Mass incarceration costs the U.S. at least $182 billion every year, with over $450 billion spent to incarcerate individuals on drug charges in federal prisons.
157.
International Monetary Fund.
IMF fossil fuel subsidies data: 2023 update. (2023)
Globally, fossil fuel subsidies were $7 trillion in 2022 or 7.1 percent of GDP. The United States subsidies totaled $649 billion. Underpricing for local air pollution costs and climate damages are the largest contributor, accounting for about 30 percent each.
158.
Papanicolas, Irene et al. Health care spending in the united states and other high-income countries.
Papanicolas et al. https://jamanetwork.com/journals/jama/article-abstract/2674671 (2018)
The US spent approximately twice as much as other high-income countries on medical care (mean per capita: $9,892 vs $5,289), with similar utilization but much higher prices. Administrative costs accounted for 8% of US spending vs 1-3% in other countries. US spending on pharmaceuticals was $1,443 per capita vs $749 elsewhere. Despite spending more, US health outcomes are not better. Additional sources: https://jamanetwork.com/journals/jama/article-abstract/2674671
.
159.
Hsieh, C.-T. & Moretti, E. Housing constraints and spatial misallocation.
American Economic Journal: Macroeconomics https://www.aeaweb.org/articles?id=10.1257/mac.20170388 (2019)
We quantify the amount of spatial misallocation of labor across US cities and its aggregate costs. Tight land-use restrictions in high-productivity cities like New York, San Francisco, and Boston lowered aggregate US growth by 36% from 1964 to 2009. Local constraints on housing supply have had enormous effects on the national economy. Additional sources: https://www.aeaweb.org/articles?id=10.1257/mac.20170388
.
161.
Tax Foundation. Tax compliance costs the US economy $546 billion annually.
https://taxfoundation.org/data/all/federal/irs-tax-compliance-costs/ (2024)
Americans will spend over 7.9 billion hours complying with IRS tax filing and reporting requirements in 2024. This costs the economy roughly $413 billion in lost productivity. In addition, the IRS estimates that Americans spend roughly $133 billion annually in out-of-pocket costs, bringing the total compliance costs to $546 billion, or nearly 2 percent of GDP.
162.
Cook, C., Cole, G., Asaria, P., Jabbour, R. & Francis, D. P. Annual global economic burden of heart disease.
International Journal of Cardiology https://www.internationaljournalofcardiology.com/article/S0167-5273(13)02238-9/abstract (2014)
Heart failure alone: $108 billion/year (2012 global analysis, 197 countries) US CVD: $555B (2016) → projected $1.8T by 2050 LMICs total CVD loss: $3.7T cumulative (2011-2015, 5-year period) CVD is costliest disease category in most developed nations Note: No single $2.1T global figure found; estimates vary widely by scope and year Additional sources: https://www.ahajournals.org/doi/10.1161/CIR.0000000000001258
.
163.
Source: US Life Expectancy FDA Budget 1543-2019 CSV.
US life expectancy growth 1880-1960: 3.82 years per decade. (2019)
Pre-1962: 3.82 years/decade Post-1962: 1.54 years/decade Reduction: 60% decline in life expectancy growth rate Additional sources: https://ourworldindata.org/life-expectancy | https://www.mortality.org/ | https://www.cdc.gov/nchs/nvss/mortality_tables.htm
.
164.
Source: US Life Expectancy FDA Budget 1543-2019 CSV.
Post-1962 slowdown in life expectancy gains. (2019)
Pre-1962 (1880-1960): 3.82 years/decade Post-1962 (1962-2019): 1.54 years/decade Reduction: 60% decline Temporal correlation: Slowdown occurred immediately after 1962 Kefauver-Harris Amendment Additional sources: https://ourworldindata.org/life-expectancy | https://www.mortality.org/ | https://www.cdc.gov/nchs/nvss/mortality_tables.htm
.
165.
Centers for Disease Control and Prevention.
US life expectancy 2023. (2024)
US life expectancy at birth was 77.5 years in 2023 Male life expectancy: 74.8 years Female life expectancy: 80.2 years This is 6-7 years lower than peer developed nations despite higher healthcare spending Additional sources: https://www.cdc.gov/nchs/fastats/life-expectancy.htm
.
166.
US Census Bureau.
US median household income 2023. (2024)
US median household income was $77,500 in 2023 Real median household income declined 0.8% from 2022 Gini index: 0.467 (income inequality measure) Additional sources: https://www.census.gov/library/publications/2024/demo/p60-282.html
.
167.
Manuel, D. U.s. Defense spending history: 100 years of military budgets.
DaveManuel.com https://www.davemanuel.com/us-defense-spending-history-military-budget-data.php (2025)
US military spending in constant 2024 dollars: 1939 $29B (pre-WW2 baseline), 1940 $37B, 1944 $1,383B, 1945 $1,420B (peak), 1946 $674B, 1947 $176B, 1948 $117B, 2024 $886B. The post-WW2 demobilization cut spending 88% in two years (1945-1947). Current peacetime spending ($886B) is 30x the pre-WW2 baseline and 62% of peak WW2 spending, in inflation-adjusted dollars.
168.
Statista. US military budget as percentage of GDP.
Statista https://www.statista.com/statistics/262742/countries-with-the-highest-military-spending/ (2024)
U.S. military spending amounted to 3.5% of GDP in 2024. In 2024, the U.S. spent nearly $1 trillion on its military budget, equal to 3.4% of GDP. Additional sources: https://www.statista.com/statistics/262742/countries-with-the-highest-military-spending/ | https://www.sipri.org/sites/default/files/2025-04/2504_fs_milex_2024.pdf
.
169.
US Census Bureau. Number of registered or eligible voters in the u.s.
US Census Bureau https://www.census.gov/newsroom/press-releases/2025/2024-presidential-election-voting-registration-tables.html (2024)
73.6% (or 174 million people) of the citizen voting-age population was registered to vote in 2024 (Census Bureau). More than 211 million citizens were active registered voters (86.6% of citizen voting age population) according to the Election Assistance Commission. Additional sources: https://www.census.gov/newsroom/press-releases/2025/2024-presidential-election-voting-registration-tables.html | https://www.eac.gov/news/2025/06/30/us-election-assistance-commission-releases-2024-election-administration-and-voting
.
170.
U.S. Senate. Treaties.
U.S. Senate https://www.senate.gov/about/powers-procedures/treaties.htm The Constitution provides that the president ’shall have Power, by and with the Advice and Consent of the Senate, to make Treaties, provided two-thirds of the Senators present concur’ (Article II, section 2). Treaties are formal agreements with foreign nations that require two-thirds Senate approval. 67 senators (two-thirds of 100) must vote to ratify a treaty for it to take effect. Additional sources: https://www.senate.gov/about/powers-procedures/treaties.htm
.
172.
Federal Election Commission.
Statistical summary of 24-month campaign activity of the 2023-2024 election cycle. (2023)
Presidential candidates raised $2 billion; House and Senate candidates raised $3.8 billion and spent $3.7 billion; PACs raised $15.7 billion and spent $15.5 billion. Total federal campaign spending approximately $20 billion. Additional sources: https://www.fec.gov/updates/statistical-summary-of-24-month-campaign-activity-of-the-2023-2024-election-cycle/
.
173.
OpenSecrets.
Federal lobbying hit record $4.4 billion in 2024. (2024)
Total federal lobbying reached record $4.4 billion in 2024. The $150 million increase in lobbying continues an upward trend that began in 2016. Additional sources: https://www.opensecrets.org/news/2025/02/federal-lobbying-set-new-record-in-2024/
.
174.
Columbia/NBER. Odds of a single vote being decisive in a u.s. Presidential election.
Columbia/NBER: What Is the Probability Your Vote Will Make a Difference? https://sites.stat.columbia.edu/gelman/research/published/probdecisive2.pdf (2012)
National average: 1 in 60 million chance (2008 election analysis by Gelman, Silver, Edlin) Swing states (NM, VA, NH, CO): 1 in 10 million chance Non-competitive states: 34 states >1 in 100 million odds; 20 states >1 in 1 billion Washington DC: 1 in 490 billion odds Methodology: Probability state is necessary for electoral college win × probability state vote is tied Additional sources: https://sites.stat.columbia.edu/gelman/research/published/probdecisive2.pdf | https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1465-7295.2010.00272.x
.
175.
Hutchinson and Kirk.
Valley of death in drug development. (2011)
The overall failure rate of drugs that passed into Phase 1 trials to final approval is 90%. This lack of translation from promising preclinical findings to success in human trials is known as the "valley of death." Estimated 30-50% of promising compounds never proceed to Phase 2/3 trials primarily due to funding barriers rather than scientific failure. The late-stage attrition rate for oncology drugs is as high as 70% in Phase II and 59% in Phase III trials.
176.
DOT. DOT value of statistical life ($13.6M).
DOT: VSL Guidance 2024 https://www.transportation.gov/office-policy/transportation-policy/revised-departmental-guidance-on-valuation-of-a-statistical-life-in-economic-analysis (2024)
Current VSL (2024): $13.7 million (updated from $13.6M) Used in cost-benefit analyses for transportation regulations and infrastructure Methodology updated in 2013 guidance, adjusted annually for inflation and real income VSL represents aggregate willingness to pay for safety improvements that reduce fatalities by one Note: DOT has published VSL guidance periodically since 1993. Current $13.7M reflects 2024 inflation/income adjustments Additional sources: https://www.transportation.gov/office-policy/transportation-policy/revised-departmental-guidance-on-valuation-of-a-statistical-life-in-economic-analysis | https://www.transportation.gov/regulations/economic-values-used-in-analysis
.
177.
PLOS ONE. Cost per DALY for vitamin a supplementation.
PLOS ONE: Cost-effectiveness of "Golden Mustard" for Treating Vitamin A Deficiency in India (2010) https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0012046 (2010)
India: $23-$50 per DALY averted (least costly intervention, $1,000-$6,100 per death averted) Sub-Saharan Africa (2022): $220-$860 per DALY (Burkina Faso: $220, Kenya: $550, Nigeria: $860) WHO estimates for Africa: $40 per DALY for fortification, $255 for supplementation Uganda fortification: $18-$82 per DALY (oil: $18, sugar: $82) Note: Wide variation reflects differences in baseline VAD prevalence, coverage levels, and whether intervention is supplementation or fortification Additional sources: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0012046 | https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0266495
.
180.
PMC. Cost-effectiveness threshold ($50,000/QALY).
PMC https://pmc.ncbi.nlm.nih.gov/articles/PMC5193154/ The $50,000/QALY threshold is widely used in US health economics literature, originating from dialysis cost benchmarks in the 1980s. In US cost-utility analyses, 77.5% of authors use either $50,000 or $100,000 per QALY as reference points. Most successful health programs cost $3,000-10,000 per QALY. WHO-CHOICE uses GDP per capita multiples (1× GDP/capita = "very cost-effective", 3× GDP/capita = "cost-effective"), which for the US ( $70,000 GDP/capita) translates to $70,000-$210,000/QALY thresholds. Additional sources: https://pmc.ncbi.nlm.nih.gov/articles/PMC5193154/ | https://pmc.ncbi.nlm.nih.gov/articles/PMC9278384/
.
181.
Integrated Benefits Institute. Chronic illness workforce productivity loss.
Integrated Benefits Institute 2024 https://www.ibiweb.org/resources/chronic-conditions-in-the-us-workforce-prevalence-trends-and-productivity-impacts (2024)
78.4% of U.S. employees have at least one chronic condition (7% increase since 2021) 58% of employees report physical chronic health conditions 28% of all employees experience productivity loss due to chronic conditions Average productivity loss: $4,798 per employee per year Employees with 3+ chronic conditions miss 7.8 days annually vs 2.2 days for those without Note: 28% productivity loss translates to roughly 11 hours per week (28% of 40-hour workweek) Additional sources: https://www.ibiweb.org/resources/chronic-conditions-in-the-us-workforce-prevalence-trends-and-productivity-impacts | https://www.onemedical.com/mediacenter/study-finds-more-than-half-of-employees-are-living-with-chronic-conditions-including-1-in-3-gen-z-and-millennial-employees/ | https://debeaumont.org/news/2025/poll-the-toll-of-chronic-health-conditions-on-employees-and-workplaces/
.
182.
Drugs.com. Time to develop one drug: 17 years.
Drugs.com https://www.drugs.com/fda-approval-process.html Time to develop one drug: 17 years Additional sources: https://www.drugs.com/fda-approval-process.html | https://www.fdareview.org/issues/the-drug-development-and-approval-process/
.
183.
GovTrack. Kefauver harris amendment of 1962.
GovTrack https://www.govtrack.us/congress/bills/87/s1552 (1962)
regulating efficacy testing via the 1962 Kefauver Harris Amendment. The 1962 regulations made these large real-world efficacy trials illegal. Additional sources: https://www.govtrack.us/congress/bills/87/s1552 | https://www.fda.gov/about-fda/histories-product-regulation/promoting-safe-effective-drugs-100-years | https://pmc.ncbi.nlm.nih.gov/articles/PMC4101807/
.
184.
FEE. Patients and doctors vs FDA.
FEE: Patients and Doctors vs FDA https://fee.org/articles/patients-and-doctors-vs-the-fda Dale Gieringer (1985): 21,000-120,000 lives lost per decade from FDA delay Gieringer: "Loss of life from delay alone in the hundreds of thousands" (not millions) Beta-blockers alone: William Wardell estimated "10,000 lives/year" if allowed; FDA delay 1965-1976 Sam Peltzman: Post-1962 death toll from regulatory delay "easily number in thousands per year Practolol (beta-blocker): "Could save 10,000 lives/year" (Wardell estimate) FDA allowed propranolol 1968 (3 years after Europe); for hypertension/angina not until 1978 Note: "4-10 million" figure not found in sources. Gieringer’s estimates: 21K-120K deaths per decade, "hundreds of thousands" total (not millions). Specific drug delays (beta-blockers): 100K deaths estimated Additional sources: https://fee.org/articles/patients-and-doctors-vs-the-fda | https://www.econlib.org/library/Enc/DrugLag.html | https://www.fdareview.org/issues/theory-evidence-and-examples-of-fda-harm/
.
185.
FDAReview.org. Estimated deaths due to FDA delay in approving beta blockers.
FDAReview.org: FDA Harm https://www.fdareview.org/issues/theory-evidence-and-examples-of-fda-harm/ (2011)
Beta blockers approved in Europe mid-1970s, FDA didn’t approve until 1981 FDA estimated the drug could save 17,000 lives/year after approval Estimated 100,000 deaths from secondary heart attacks during 6-7 year delay Note: FDA imposed moratorium due to possible animal carcinogenicity despite human clinical evidence from 1974 Additional sources: https://www.fdareview.org/issues/theory-evidence-and-examples-of-fda-harm/ | https://www.ocregister.com/2011/02/09/walter-williams-death-by-fda-delay-denials/
.
186.
CRS. Evidence of FDA regulatory capture by pharmaceutical industry.
CRS: FDA Human Medical Product User Fees https://www.congress.gov/crs-product/R44750 FY2023: User fees = 75% of PDUFA program costs (vs 7% in FY1993) FY2022: User fees = 66% ($1.4B) of human drugs program budget FY2022: User fees = 46% ($2.9B) of FDA’s total $6.2B budget Pharma finances 75% of FDA’s drug division (New York Times) Concerns: Budgetary dependence, urgency of PDUFA reauthorizations, required industry participation in negotiations Most PDUFA policy changes favor industry: decreased regulatory standards, shorter approval times, increased industry involvement FDA maintains decisions based on science, not fee collection ability Note: Regulatory capture concerns well-documented. Industry funding grew from 7% (1993) to 75% (2023) of drug review program. No evidence found for "10x government salary" claim for FDA reviewers Additional sources: https://www.congress.gov/crs-product/R44750 | https://aspe.hhs.gov/sites/default/files/documents/e4a7910607c0dd76c40aa61151d154f9/FDA-User-Fee-Issue-Brief.pdf | https://pmc.ncbi.nlm.nih.gov/articles/PMC8917050/
.
198.
Sakaeda et al. FAERS adverse event underreporting rate.
PubMed: Empirical estimation of under-reporting in FAERS https://pubmed.ncbi.nlm.nih.gov/28447485/ (2017)
Empirical estimation: Average reporting rate approximately 6%, meaning 94% of adverse events are underreported Variability: 0.01% to 44% for statin events; 0.002% to >100% for biological drugs; 20% to >100% for narrow therapeutic index (NTI) drugs Selective reporting: Serious, unusual events more likely reported than mild or expected ones Newly marketed drugs: Higher reporting rates due to heightened awareness Older drugs: Events often under-reported Note: FAERS voluntary reporting system captures only "tip of the iceberg" of drug safety problems. Under-reporting introduces inherent biases and limitations in pharmacovigilance data Additional sources: https://pubmed.ncbi.nlm.nih.gov/28447485/ | https://pmc.ncbi.nlm.nih.gov/articles/PMC12393772/
.
199.
Graham, David (FDA) | Lancet. Vioxx cardiovascular deaths (rofecoxib).
PMC: FDA incapable of protecting against another Vioxx https://pmc.ncbi.nlm.nih.gov/articles/PMC534432/ (2007)
Graham testimony (2004): 88,000-139,000 U.S. heart attacks/strokes from Vioxx; up to 55,000 deaths (40% fatality rate) Lancet study estimate: 88,000 Americans had heart attacks from Vioxx; 38,000 died FDA memo (2004): Vioxx contributed to 27,785 heart attacks and sudden cardiac deaths (1999-2003) High-dose Vioxx: Tripled risk of heart attacks and sudden cardiac death Prescriptions: 92.8 million U.S. prescriptions 1999-2003 Withdrawn: September 30, 2004 after APPROVE trial showed cardiovascular risks Note: Vioxx case demonstrates failure of passive post-market surveillance (FAERS) to detect safety signals in time. Voluntary reporting missed cardiovascular risks for years despite millions of prescriptions Additional sources: https://pmc.ncbi.nlm.nih.gov/articles/PMC534432/ | https://www.npr.org/2007/11/10/5470430/timeline-the-rise-and-fall-of-vioxx | https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(05)67712-4/fulltext
.
200.
Sinn, M. P.
The Continuous Evidence Generation Protocol: Two-Stage Validation (RWE → Pragmatic Trials).
https://manual.warondisease.org/knowledge/appendix/dfda-spec-paper.html (2025) doi:
10.5281/zenodo.18203375 We present the Predictor Impact Score (PIS), a novel composite metric operationalizing Bradford Hill causality criteria for automated signal detection from aggregated N-of-1 observational studies. Combined with pragmatic trial confirmation (based on evidence from 108+ embedded trials), this two-stage framework would generate validated outcome labels at 44.1x lower cost than traditional Phase III trials. This enables continuous, population-scale pharmacovigilance and precision dosing recommendations.
201.
Sinn, M. P.
Ubiquitous Pragmatic Trial Impact Analysis: How to Prevent a Year of Death and Suffering for 84 Cents.
https://manual.warondisease.org/knowledge/appendix/dfda-impact-paper.html (2025) doi:
10.5281/zenodo.18243914 Only 15 diseases/year get their first treatment each year. With 6.65 thousand diseases lacking effective treatments, the backlog would take 443 years to clear. Integrating pragmatic trials into standard healthcare increases trial capacity 12.3x, cutting that timeline from 443 years to 36 years. The average untreated disease gets a treatment 212 years earlier, saving 10.7 billion deaths at $0.842 per year of healthy life saved.
202.
AllTrials. Publication rate of clinical trial results.
AllTrials: Half of Trials Unreported https://www.alltrials.net/news/half-of-all-trials-unreported/ (2013)
50.0% of clinical trials never publish results (NHS-funded systematic review, 2010) Schmucker et al (2014): 53% of trials published (analyzing 39 studies, >20,000 trials) Munch et al (2014): 46% of pain treatment trials published Chang et al (2015): 49% of high-risk cardiac device trials published Positive findings: 3X more likely to be published than negative results Antidepressant example: Published literature showed 94% positive trials; FDA analysis showed only 51% positive Additional sources: https://www.alltrials.net/news/half-of-all-trials-unreported/ | https://www.nature.com/articles/nature.2013.14286 | https://pmc.ncbi.nlm.nih.gov/articles/PMC8276556/
.
203.
Science/AAAS. Estimated annual cost of repeating failed experiments due to non-publication of results.
Science/AAAS https://www.science.org/content/article/study-claims-28-billion-year-spent-irreproducible-biomedical-research (2020)
Up to 50.0% of published preclinical research is irreproducible, with an estimated annual cost of $28 billion in the U.S. alone. This is based on $56B annual spending on preclinical research × 50.0% irreproducibility rate. Main causes: reagents/materials (36%), study design (28%), data analysis (25%), protocols (11%). Additional sources: https://www.science.org/content/article/study-claims-28-billion-year-spent-irreproducible-biomedical-research | https://www.idbs.com/2020/11/replicating-science-28-billion-is-wasted-every-year-in-the-us-alone/
.
204.
FDLI. Japan’s regenerative medicine act and conditional approval pathway.
FDLI: Japan’s Regen Med Pathways https://www.fdli.org/2019/02/global-focus-japans-regenerative-medicine-regulatory-pathways-encouraging-innovation-and-patient-access/ (2019)
Act on Safety of Regenerative Medicine (RM Act) + amended Pharmaceuticals and Medical Devices Act (PMD Act): passed Nov 2013, effective Nov 2014 Conditional and time-limited approval pathway: Obtain approval after exploratory trials demonstrate probable benefit and proven safety 7-year conditional approval period to confirm clinical benefit (e.g., using surrogate endpoints) SAKIGAKE designation (April 2015): Expedited pathway for innovative products targeting serious/life-threatening diseases without effective treatment Benefits: Prioritized consultation, accelerated review, extended re-examination period, premium pricing Examples: Terumo’s HeartSheet and Stemirac obtained conditional approval; Stemirac also SAKIGAKE-designated Additional sources: https://www.fdli.org/2019/02/global-focus-japans-regenerative-medicine-regulatory-pathways-encouraging-innovation-and-patient-access/ | https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/310/Experiences-from-Japan-SAKIGAKE-Designation-System-for-Regenerative-Medical-Products | https://pmc.ncbi.nlm.nih.gov/articles/PMC6696404/
.
205.
EMA. EU compassionate use program for experimental drugs.
EMA: Compassionate Use https://www.ema.europa.eu/en/human-regulatory-overview/research-development/compassionate-use Established by: Article 83 of Regulation (EC) No 726/2004 Eligibility: Life-threatening, long-lasting, or seriously debilitating illnesses Requires: No satisfactory authorized treatment; medicine in trials or approval process EMA role: CHMP provides recommendations; national authorities implement programs Pan-European programs rare: Only 6 approved by EMA in last 10 years vs hundreds of national programs Each EU member state sets own rules and procedures Note: Pan-European framework exists but rarely used due to complex navigation across varying national regulations. Most programs remain country-specific Additional sources: https://www.ema.europa.eu/en/human-regulatory-overview/research-development/compassionate-use | https://pmc.ncbi.nlm.nih.gov/articles/PMC5116859/ | https://www.eurordis.org/information-support/compassionate-use/
.
207.
PMC. Number of patients helped by the u.s. Right to try act.
PMC: Understanding Right to Try https://pmc.ncbi.nlm.nih.gov/articles/PMC7416898/ (2024)
2018-2022: Supported access to only 12 products total; 4 products in 2023 Number of people treated: Not publicly reported, "likely only in the hundreds Specific documented cases: 75+ neuroendocrine cancer patients (LU-177); at least 1 ALS patient (NurOwn); 7 glioblastoma patients (Gliovac) Minimal safety reporting: Only annual basis, no requirement to publish results/outcomes Additional sources: https://pmc.ncbi.nlm.nih.gov/articles/PMC7416898/ | https://www.statnews.com/2024/08/02/trump-gave-patients-right-to-try-it-hasnt-helped-them/ | https://www.healio.com/news/hematology-oncology/20200303/right-to-try-a-wellintentioned-but-misguided-law
.
208.
CNN. FDA delay in approving rapid COVID-19 tests.
CNN: How Government Delayed Testing https://www.cnn.com/2020/04/09/politics/coronavirus-testing-cdc-fda-red-tape-invs/index.html (2020)
Feb 16, 2020: Seattle research lab ordered to stop COVID testing without FDA approval May 2020: Gates Foundation partnership instructed to discontinue testing until authorization Feb 29, 2020: U. Nebraska finally got FDA permission after Feb 4 special permission Timeline: Critical delays measured in weeks-to-months (Feb-March 2020), not specific "6 months FDA guidance suggested EUAs needed for laboratory-developed tests (LDTs), causing delays By May 2020: 400+ test applications awaiting FDA review Note: Delays were weeks-to-months during critical Feb-March 2020 period, not continuous "6 months." FDA intervention added minimal value while contributing to deadly delays Additional sources: https://www.cnn.com/2020/04/09/politics/coronavirus-testing-cdc-fda-red-tape-invs/index.html | https://www.yalelawjournal.org/forum/deadly-delay-the-fdas-role-in-americas-covid-testing-debacle | https://oig.hhs.gov/oei/reports/OEI-01-20-00380.asp
.
209.
NPR. Contamination of early CDC COVID-19 tests in 2020.
NPR: CDC Test Flawed https://www.npr.org/2020/11/06/929078678/cdc-report-officials-knew-coronavirus-test-was-flawed-but-released-it-anyway (2020)
CDC distributed flawed test kits Feb 6, 2020 - contaminated reagents caused false positives 24 of 26 public health labs found contamination, CDC recalled kits by Feb 10 Tests made in CDC lab (not manufacturing facility), violated sound manufacturing practices Contamination occurred in Respiratory Virus Diagnostic Lab during processing Note: Delays had significant consequences for early pandemic tracking and response Additional sources: https://www.npr.org/2020/11/06/929078678/cdc-report-officials-knew-coronavirus-test-was-flawed-but-released-it-anyway | https://www.cnn.com/2020/04/18/politics/cdc-coronavirus-testing-contamination/index.html
.
210.
PMC. Estimated excess deaths attributed to FDA’s COVID-19 response.
PMC: EUAs vs FDA Approval Implications https://pmc.ncbi.nlm.nih.gov/articles/PMC8101583/ FDA regulations restricted clinician/patient access to COVID-19 testing, remdesivir, vaccines General drug delay estimate: 37,000-76,000 deaths per one-year delay Testing delays: By May 2020, 400+ applications awaiting FDA review Seattle lab ordered to stop testing Feb 16 for lack of FDA approval Gates Foundation partnership instructed to discontinue testing May 2020 until authorization EUA process prevented months of vaccine/testing delays Note: Specific "500,000+" figure not found in sources. Research shows FDA testing restrictions caused weeks-to-months of critical delays (Feb-March 2020). One-year drug delay = 37-76K deaths Additional sources: https://pmc.ncbi.nlm.nih.gov/articles/PMC8101583/ | https://pmc.ncbi.nlm.nih.gov/articles/PMC8012986/ | https://www.yalelawjournal.org/forum/deadly-delay-the-fdas-role-in-americas-covid-testing-debacle
.